The International Myeloma Foundation (IMF) today announced the publication in The Lancet of the results of a randomized, phase III trial, conducted by SWOG, a publicly funded international cancer clinical trials network, and led by IMF chairman of the board Brian G.M. Durie, MD. This important trial compared the effectiveness of two drug regimens in patients undergoing their first round of treatment for multiple myeloma. The trial shows that a three-drug combination - known as VRd - delays recurrence and lengthens life for myeloma patients, indicating a possible new standard of care.
One regimen used in the study was lenalidomide with dexamethasone, a standard first-line treatment for myeloma patients. The other drug regimen also included bortezomib, a second-line drug typically given to myeloma patients whose cancer progresses after initial therapy. SWOG researchers found that the addition of bortezomib earlier made a difference for myeloma patients, giving them about another year of remission and another year of life compared to the standard two-drug regimen.
"Our results are clear. Using bortezomib in combination with lenalidomide and dexamethasone in front-line treatment - hitting the disease early and hard - makes a meaningful difference for myeloma patients," said study principal investigator Dr. Durie, a physician at Cedars-Sinai Outpatient Cancer Center in Los Angeles. "Our results represent a potential new standard of care."
"This is a landmark study that lends clarity to frontline therapy of myeloma," said Dr. S. Vincent Rajkumar of Mayo Clinic and a co-author of the study. "Newer alternatives to VRd may be more expensive, cumbersome, or toxic. These regimens will therefore need to show superiority over VRd in randomized trials."
Also worth noting, Dr. Rajkumar said, is that the VRd regimen will become even more cost effective as the drugs in this combination become generic over time.
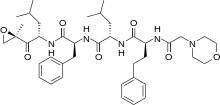
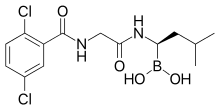
 bortezomib
bortezomib  lenalidomide
lenalidomide
 dexamethasone
dexamethasone
Patients in the trial receiving bortezomib, along with lenalidomide and dexamethasone, in their first six months of treatment had a median remission time of 43 months compared to a median remission of 30 months for patients who received lenalidomide and dexamethasone alone. Researchers also found that patients who received bortezomib lived a median of 75 months, or about six years, after their initial treatment. Patients who received the standard two-drug treatment lived a median of 64 months, or about five years, after initial treatment.
Celebrating its 60th year, SWOG has conducted more than 1,300 cancer trials that have led to FDA approval of 14 new drugs and led to more than 100 changes to cancer standards of care. SWOG is part of the NCI's National Clinical Trials Network (NCTN), the nation's largest and oldest publicly funded cancer research network. SWOG members and other members in the NCTN enrolled 471 eligible and consented adult patients in the study, known as S0777, between February 2008 and February 2012, at 139 institutions across the US.
Patients ranged in age from 28 to 87, had active myeloma, and had not had a stem-cell transplant or any prior treatment for their disease. Patients were randomized into two groups. One group received the standard two-drug treatment for six cycles over six months. That includes lenalidomide, an immunomodulating therapy marketed as Revlimid by Celegene Corporation. The other group received a three-drug combination that included bortezomib, a proteasome inhibitor marketed as Velcade by Millennium Pharmaceuticals. These patients received the triple combination therapy for eight cycles over six months.