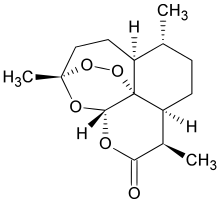
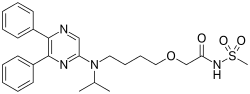
We know that, Omigapil (TCH346 or CGP3466) is a drug that was developed by Novartis and tested in clinical trials for its ability to help treat Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS). The development for PD and ALS have been terminated due to lack of benefit, but Santhera Pharmaceuticals bought the compound for development for the treatment of congenital muscular dystrophy (CMD). Omigapil was first synthesized at Ciba-Geigy, Basel, Switzerland. Santhera Pharmaceuticals has since taken over production of omigapil and preclinical trials for CMD. In May 2008, omigapil was granted orphan designation to commence clinical trials for. Pharmacokinetic trials are scheduled to commence enrollment in the second half of 2012 to determine the appropriate pharmacokinetic profile of the drug for children with laminin-α2-deficient congenital muscular dystrophy (MDC1A) and collagen VI related myopathy. Santhera Pharmaceuticals will use the phase 1 clinical trial to determine if the drug is safe and acts with the same pharmacokinetic profile in children as it does in adults. The impending clinical trial will take place in the United States at the National Institute of Neurological Disorders and Stroke/National Institute of Health(NNDCS/NINDS) (Bethesda, Maryland) and in the United Kingdom at Great Ormond Street Hospital (UCL
The compound CGP3466B, already proven nontoxic for people, may effectively and rapidly treat depression, according to results of a study in mice. The Johns Hopkins Medicine neuroscientists who conducted the research say that the compound -- previously shown to block cocaine craving in the brains of rodents -- delivers antidepressant effects to mice within hours instead of weeks or months, like currently available antidepressants. The results of the study will be summarized Jan. 12 online in the journal Molecular Psychiatry.
"One of the promising things about CGP3466B is that it targets a new network of proteins," says Solomon Snyder, M.D., professor of neuroscience at the Johns Hopkins University School of Medicine. "That means it may work in patients who are unresponsive to other types of drugs and it lays the foundation for the development of a new class of fast-acting antidepressants that target the same network."
The team's discovery came out of investigations into the workings of a different drug, ketamine, long used primarily at high doses to induce anesthesia during surgery but known, at lower doses, to be a fast-acting antidepressant. Unfortunately, Snyder says, ketamine is addictive and can produce schizophrenialike symptoms, making it unsuitable for prolonged use, but his team hoped it could shed light on how to make a better fast-acting antidepressant.