The pyran ring is present in so many useful compounds, such as pharmaceuticals (antibiotics, anti-infectives, cardiovascular agents, neurological modulators, anti-allergic, anti-asthmatic, anti-inflammatory agents, reproductive and genitourinary agents, growth promoters and antidiabetic agents), veterinary products, agrochemicals, toxins, polymers and additives, photosensitizers and photoinitiators, surfactants, food products, dyes and pigments. This fact keeps motivating synthetic organic chemists to develop newer facile synthetic methods to make these compounds accessible in high enantiomeric purity. Bansal and co-workers have recently succeeded in obtaining a series of phenyl substituted 3,6-Dihydro-2H-pyran derivatives in 68 to 95% enantiomeric excess.
This report has two important features. It illustrates that differently substituted 2-phenyl-3,6-dihydro-2H-pyrans can be obtained in high yield with high enantiomeric purity involving relatively simple experimental method. Secondly, the enantiomeric excess has been rationalized on the basis of computational calculations at the DFT level. It is hoped that the results would be useful for further research in this field.
 (Dapsone)
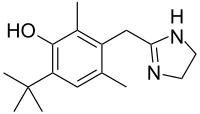
(Dapsone) Palbociclib
Palbociclib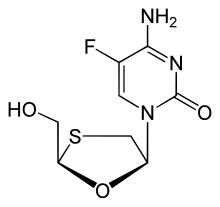 Emtricitabine
Emtricitabine  Rilpivirine
Rilpivirine Tenofovir alafenamide
Tenofovir alafenamide


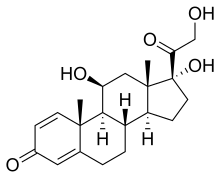 (Prednisole)
(Prednisole) (Indomethacin)
(Indomethacin)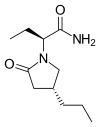
 (
( (Donepezil)
(Donepezil) 

 (Elbasvir)
(Elbasvir)  (Grazoprevir)
(Grazoprevir)
 Tetracaine
Tetracaine