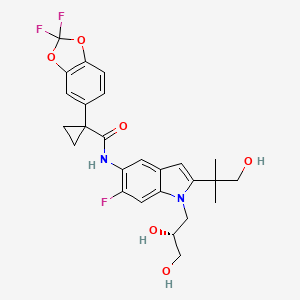
The U.S. Food and Drug Administration, has approved Erleada (apalutamide) for the treatment of patients with prostate cancer that has not spread (non-metastatic), but that continues to grow despite treatment with hormone therapy (castration-resistant). This is the first FDA-approved treatment for non-metastatic, castration-resistant prostate cancer.
“The FDA evaluates a variety of methods that measure a drug’s effect, called endpoints, in the approval of oncology drugs. This approval is the first to use the endpoint of metastasis-free survival, measuring the length of time that tumors did not spread to other parts of the body or that death occurred after starting treatment,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “In the trial supporting approval, Erleada had a robust effect on this endpoint. This demonstrates the agency’s commitment to using novel endpoints to expedite important therapies to the American public."
According to the National Cancer Institute (NCI) at the National Institutes of Health, prostate cancer is the second most common form of cancer in men in the U.S.. The NCI estimates approximately 161,360 men were diagnosed with prostate cancer in 2017, and 26,730 were expected to die of the disease. Approximately 10 to 20 percent of prostate cancer cases are castration-resistant, and up to 16 percent of these patients show no evidence that the cancer has spread at the time of the castration-resistant diagnosis.
Erleada works by blocking the effect of androgens, a type of hormone, on the tumor. These androgens, such as testosterone, can promote tumor growth.
The safety and efficacy of Erleada was based on a randomized clinical trial of 1,207 patients with non-metastatic, castration-resistant prostate cancer. Patients in the trial either received Erleada or a placebo. All patients were also treated with hormone therapy, either with gonadotropin-releasing hormone (GnRH) analog therapy or with surgery to lower the amount of testosterone in their body (surgical castration). The median metastasis-free survival for patients taking Erleada was 40.5 months compared to 16.2 months for patients taking a placebo.
Common side effects of Erleada include fatigue, high blood pressure (hypertension), rash, diarrhea, nausea, weight loss, joint pain (arthralgia), falls, hot flush, decreased appetite, fractures and swelling in the limbs (peripheral edema).
Severe side effects of Erleada include falls, fractures and seizures.
This application was granted Priority Review, under which the FDA’s goal is to take action on an application within 6 months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The sponsor for Erleada is the first participant in the FDA’s recently-announced Clinical Data Summary Pilot Program, an effort to provide stakeholders with more usable information on the clinical evidence supporting drug product approvals and more transparency into the FDA’s decision-making process. Soon after approval, certain information from the clinical summary report will post with the Erleada entry on Drugs@FDA and on the new pilot program landing page.
 Ivacafto
Ivacafto