
‘’This approval fulfills an unmet need by addressing the shortcomings and handling complexities of the current lyophilized powder formulation,” said Sharon Cunningham, Chief Executive Officer and Co-Founder of Shorla Oncology. “We have taken a vital oncology drug and made it easier for oncology clinics and hospitals to use, while also reducing medical personnel exposure to a hazardous drug.”
Tepylute, formerly SH-105, is the third FDA-approved drug for Shorla, and a significant milestone for the company as it seeks approval for several cancer-fighting drugs for the U.S. market.
“The approval of Tepylute represents an important milestone for Shorla as our first in-house developed NDA,” said Orlaith Ryan, Chief Technical Officer and Co-Founder of Shorla Oncology.
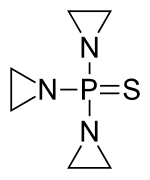
Tepylute is a liquid form of a well-established, standard of care oncology drug, thiotepa. The new formulation eliminates the need for complex and time-consuming reconstitution. It provides consistent dosing accuracy and allows for “just in time” preparation.2
“Among Tepylute’s many benefits, it removes the necessity to reconstitute which can introduce additional risks of drug preparation errors,” emphasized Rayna Herman, Chief Commercial Officer. “We look forward to providing an update on our launch plans for Tepylute in the near future.”
The American Cancer Society estimates that more than 300,000 women will be diagnosed with breast cancer in the U.S in 2024.3 About 19,680 women will be diagnosed with ovarian cancer in the United States.4
Shorla Oncology is currently marketing two products with a robust pipeline including SH-201, the first palatable oral liquid treatment for certain forms of leukemia and other cancers. In April, the company announced the FDA had accepted SH-201 for an NDA review with an expected action date of November 30, 2024.
‘’This approval fulfills an unmet need by addressing the shortcomings and handling complexities of the current lyophilized powder formulation,” said Sharon Cunningham, Chief Executive Officer and Co-Founder of Shorla Oncology. “We have taken a vital oncology drug and made it easier for oncology clinics and hospitals to use, while also reducing medical personnel exposure to a hazardous drug.”
Tepylute, formerly SH-105, is the third FDA-approved drug for Shorla, and a significant milestone for the company as it seeks approval for several cancer-fighting drugs for the U.S. market.
“The approval of Tepylute represents an important milestone for Shorla as our first in-house developed NDA,” said Orlaith Ryan, Chief Technical Officer and Co-Founder of Shorla Oncology.
Tepylute is a liquid form of a well-established, standard of care oncology drug, thiotepa. The new formulation eliminates the need for complex and time-consuming reconstitution. It provides consistent dosing accuracy and allows for “just in time” preparation.2
“Among Tepylute’s many benefits, it removes the necessity to reconstitute which can introduce additional risks of drug preparation errors,” emphasized Rayna Herman, Chief Commercial Officer. “We look forward to providing an update on our launch plans for Tepylute in the near future.”
The American Cancer Society estimates that more than 300,000 women will be diagnosed with breast cancer in the U.S in 2024.3 About 19,680 women will be diagnosed with ovarian cancer in the United States.4
Shorla Oncology is currently marketing two products with a robust pipeline including SH-201, the first palatable oral liquid treatment for certain forms of leukemia and other cancers. In April, the company announced the FDA had accepted SH-201 for an NDA review with an expected action date of November 30, 2024.




