A new approach to killing C. difficile that silences key bacterial genes while sparing other bacteria may provide a new way to treat the most common hospital-acquired bacterial infection in the United States, according to researchers.
While conventional antibiotics treat bacterial infections, they can also cause a condition in the colon called C. difficile infection, due to the drug killing both good and bad bacteria in the gut.
In a lab, researchers created three new antibiotics that kill C. difficile by preventing the expression of bacterial genes that are important for its survival. This approach -- called antisense therapy -- allows the drug to kill only C. difficile, unlike many antibiotics that kill multiple forms of bacteria.
"We were able to show that these drugs can zero in on and kill C. difficile bacteria while leaving other bacteria alone," said Arun Sharma, associate professor of pharmacology, Penn State College of Medicine. "We're still working to refine these drugs and make them even better, with the eventual goal of testing them clinically."
David Stewart, an associate professor of surgery at the University of Arizona who along with Sharma is a co-principal investigator on this study, said the drug works in a completely different manner than the antibiotics currently used.
"These drugs are organism specific, meaning that they target only one kind of bacteria, kind of like smart antibiotics," Stewart said. "They're precise. And that's especially important with C. difficile infections because this bacteria is uniquely, selectively advantaged to exploit ecological disturbances in the human gut."
While C. difficile is normally present in the gut, other "good" bacteria are also present, and all these bacteria contribute to a person's microbiome. When a person's microbiome is healthy and balanced, it keeps bad bacteria like C. difficile under control.
But if a patient takes an antibiotic for another condition, the antibiotic kills many different types of bacteria, including the good ones keeping C. difficile under control. This allows C. difficile to thrive, causing an infection that can result in severe gastrointestinal symptoms. Since antibiotics can contribute to C. difficile infections, the researchers said a new, alternative treatment for these infections is desirable.
"Ideally, a treatment for C. difficile would have no effect on other bacteria," Stewart said.
The researchers, who recently published their findings in the Journal of Antibiotics, said that while most antibiotics lack organismal specificity -- the ability to target just one type of organism -- antisense treatments show great potential for being able to target only specific bacteria.
"Our antisense antibiotics contain genetic material which is complementary to bacterial genetic material, so we designed our genetic material to target specific genes in C. difficile," Stewart said. "And when our genetic material binds to the bacterial genetic material, it prevents the expression of bacterial genes. And that can cause C. difficile to die."
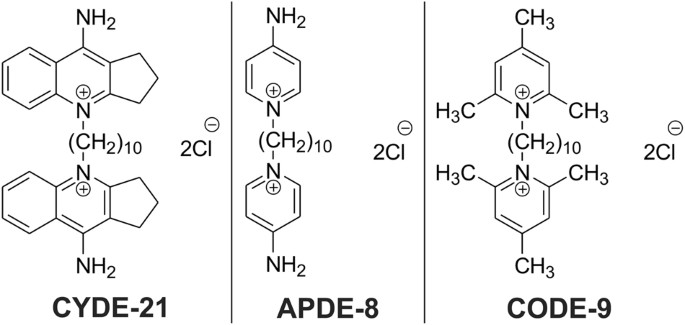
The drug tested in the study consisted of two components: the antisense compound that targeted the genetic material in C. difficile -- referred to as an antisense oligonucleotide (ASO) -- and a carrier compound that transported the ASO into the bacteria, referred to by the research team as a CAB. The researchers tested three versions of the drug, each with a different version of CAB.
The researchers tested each compound to see how much of the drug was required to kill C. difficile bacteria, whether it was toxic or not to human colon cells, and whether it also harmed other bacteria normally found in the gut -- like E. coli.
"Ultimately, we wanted these compounds to deliver the drug into the C. difficile bacteria without hurting other bacteria or the patient," Sharma said. "After testing these three, we found that one carrier in particular -- CYDE-21 -- was the best at delivering an effective dose of the drug into the bacteria."
In the future, the researchers said they will conduct further studies to continue to refine the carriers to increase their capacity and minimize their effect on other bacteria and human cells.
"In this study, as a first effort, the carrier is pretty good, and we'd like to do even better," Stewart said. "It has minimal antibacterial activity, minimal toxicity and it's an effective carrier of our cargo. So what we're working on now is modifying our carriers for future testing in preparation for animal studies."
Ref : https://www.nature.com/articles/s41429-018-0056-9




{kind=link}