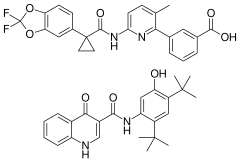
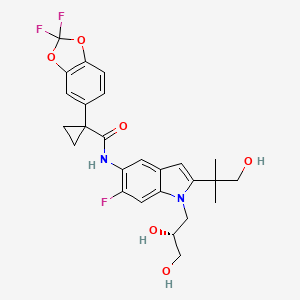
In continuation of my update on tezacaftor and ivacaftor
Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced the U.S. Food and Drug Administration (FDA) has approved Trikafta (elexacaftor/tezacaftor/ivacaftor and ivacaftor) for the treatment of cystic fibrosis (CF) in people ages 12 years and older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, the most common CF-causing mutation. With this approval, for the first time, approximately 6,000 people with CF ages 12 years and older who have one F508del mutation and one minimal function mutation (F/MF) have a medicine that targets the underlying cause of their CF. Additionally, approximately 12,000 people with one or two F508del mutations who are currently eligible for one of Vertex’s three other FDA-approved CF medicines are now also eligible for Trikafta.
“Today marks a milestone for CF patients, their families and Vertex. After a 20-year journey together, we have received FDA approval of Trikafta: a single breakthrough medicine with the potential to treat up to 90% of all people with CF in the future. For approximately 6,000 people with CF in the U.S., Trikafta is the first medicine that can treat the underlying cause of their disease,” said Jeffrey Leiden, M.D., Ph.D., Vertex's Chairman, President and Chief Executive Officer. “I want to personally thank the hundreds of Vertex scientists who have been working on this program for nearly 20 years – many of whom have dedicated their entire careers to changing the course of this disease; the CF Foundation which has provided support, encouragement and help throughout the journey; and most importantly the thousands of patients, caregivers, doctors and advocates who have courageously and persistently worked side-by-side with us to get to where we are today.”
“Today’s approval is a historic moment in cystic fibrosis care, with the potential for more people to benefit from CFTR modulator therapy to treat the basic defect of their disease,” said Steven Rowe, M.D., Director, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham. “In clinical trials, Trikafta was generally well tolerated and demonstrated improvements in multiple outcome measures in CF, including improvements in FEV1, improvements in respiratory symptoms and, in the 24-week F/MF study, a reduced rate of pulmonary exacerbations and improvements in BMI.”
“The incredible speed of this approval underscores our shared sense of urgency with the FDA and the CF community for bringing this medicine to eligible people with CF, particularly those without a medicine targeting the underlying cause of their disease,” said Reshma Kewalramani, M.D., Executive Vice President, Global Medicines Development and Medical Affairs and Chief Medical Officer at Vertex. “We remain committed to relentlessly pursuing the development of transformative therapies for all people living with this disease.”
Vertex has submitted a Marketing Authorization Application (MAA) to the European Medicines Agency (EMA) for the elexacaftor/tezacaftor/ivacaftor combination regimen. Vertex is currently evaluating elexacaftor/tezacaftor/ivacaftor in people ages 6 through 11 with F/MF and F/F CF mutations in an ongoing Phase 3 study and is committed to evaluating elexacaftor/tezacaftor/ivacaftor in children <6 years of age as part of planned future studies.


 https://en.wikipedia.org/wiki/Elexacaftor/ivacaftor/tezacaftor
https://en.wikipedia.org/wiki/Elexacaftor/ivacaftor/tezacaftor
https://en.wikipedia.org/wiki/Ivacaftor
https://en.wikipedia.org/wiki/Elexacaftor/ivacaftor/tezacaftor