
The U.S. Food and Drug Administration approved Varubi (rolapitant) to prevent delayed phase chemotherapy-induced nausea and vomiting (emesis). Varubi is approved in adults in combination with other drugs (antiemetic agents) that prevent nausea and vomiting associated with initial and repeat courses of vomit-inducing (emetogenic and highly emetogenic) cancer chemotherapy.
Nausea and vomiting are common side effects experienced by cancer patients undergoing chemotherapy. Symptoms can persist for days after the chemotherapy drugs are administered. Nausea and vomiting that occurs from 24 hours to up to 120 hours after the start of chemotherapy is referred to as delayed phase nausea and vomiting, and it can result in serious health complications. Prolonged nausea and vomiting can lead to weight loss, dehydration and malnutrition in cancer patients leading to hospitalization.
"Chemotherapy-induced nausea and vomiting remains a major issue that can disrupt patients' lives and sometimes their therapy," said Amy Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in the FDA's Center for Drug Evaluation and Research. "Today's approval provides cancer patients with another treatment option for the prevention of the delayed phase of nausea and vomiting caused by chemotherapy."
Varubi is a substance P/neurokinin-1 (NK-1) receptor antagonist. Activation of NK-1 receptors plays a central role in nausea and vomiting induced by certain cancer chemotherapies, particularly in the delayed phase. Varubi is provided to patients in tablet form.

The safety and efficacy of Varubi were established in three randomized, double-blind, controlled clinical trials where Varubi in combination with granisetron and dexamethasone was compared with a control therapy (placebo, granisetron and dexamethasone) in 2,800 patients receiving a chemotherapy regimen that included highly emetogenic (such as cisplatin and the combination of anthracycline and cyclophosphamide) and moderately emetogenic chemotherapy drugs. Those patients treated with Varubi had a greater reduction in vomiting and use of rescue medication for nausea and vomiting during the delayed phase compared to those receiving the control therapy.
Video for more info
Varubi (rolapitant) approved to prevent delayed phase chemotherapy-induced nausea and vomiting

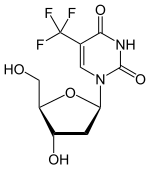 (Trifluridine)
(Trifluridine) 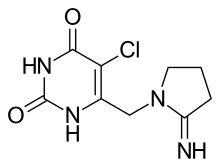 (Tipiracil)
(Tipiracil)











