

Can you give me a brief overview of bacterial lung infections and why they are so lethal?
If you trace back to the largest pandemics in human history, for example the Spanish Flu, the number of deaths was around 40 million. For flu to truly have an epidemic or pandemic potential, the lethality of the infection itself can't be so high that it doesn't allow for transmissibility. If the flu virus is too lethal than it has a harder time spreading from host to host.
Normal Lung (left) and Lung with Influenza (right). Comparative twin images of normal healthy lung in contrast to lung from a case of influenza showing microscopic disease pathology of flu. © vetpathologist / Shutterstock.com.
When scientists first started to look at what happened during the 1918-1919 pandemic, what we saw was that the major cause of death was not the flu virus but that patients died due to a secondary bacterial pneumonia. So the major question became what was the 1918 flu strain doing that caused people to become susceptible to bacterial super-infections?
So scientists began digging patient samples from the Alaskan permafrost. What they observed was that the primary flu infection caused a significant amount of lung damage in the host without killing them, which then allowed streptococcus pneumonia, the common bacterial super infection, to grow in the lung and cause systemic infection. The body becomes unable to combat this bacterial super infection, which becomes the primary cause of mortality.



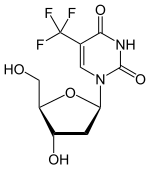 (Trifluridine)
(Trifluridine) 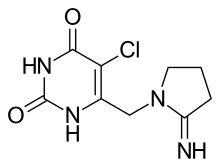 (Tipiracil)
(Tipiracil)







