
We know that, Astaxanthin /æstəˈzænθɨn/ is a keto-carotenoid. It belongs to a larger class of chemical compounds known as terpenes, which are built from five carbon precursors; isopentenyl diphosphate (or IPP) and dimethylallyl diphosphate (or DMAPP). Astaxanthin is classified as a xanthophyll (originally derived from a word meaning "yellow leaves" since yellow plant leaf pigments were the first recognized of the xanthophyll family of carotenoids), but currently employed to describe carotenoid compounds that have oxygen-containing moities, hydroxyl (-OH) or ketone (C=O), such as zeaxanthin and canthaxanthin. Indeed, astaxanthin is a metabolite of zeaxanthin and/or canthaxanthin, containing both hydroxyl and ketone functional groups. Like many carotenoids, astaxanthin is a colorful, lipid-soluble pigment. This colour is due to the extended chain of conjugated (alternating double and single) double bonds at the centre of the compound. This chain of conjugated double bonds is also responsible for the antioxidant function of astaxanthin (as well as other carotenoids) as it results in a region of decentralized electrons that can be donated to reduce a reactive oxidizing molecule.
Solix Algredients, Inc. has introduced Solasta™ Astaxanthin to the B2B ingredients marketplace.

Solasta™ is a natural astaxanthin extract produced from the microalga Haematococcus pluvialis, the richest natural source of the antioxidant. Research has shown that algal astaxanthin has superior antioxidant activity compared to other well-known antioxidants.
Solasta™ Astaxanthin is an excellent source of astaxanthin for use in dietary supplement and personal care applications. Solasta™ is non-GMO, vegetarian, and extracted in the USA. The extraction uses super-critical carbon dioxide to ensure the safety, quality, and consistency of Solasta™.
Solix Algredients is now leveraging its extensive algal cultivation expertise to supply functional ingredients for consumer end-use. "The launch of Solasta™ Astaxanthin coincides with the repositioning of Solix Algredients as a global, B2B supplier of high quality natural ingredients derived from algae," explained James Tuan, Chief Commercial Officer.
Solasta™ will be sold to manufacturers and marketers of dietary supplement and personal care products. "Solasta™ Astaxanthin is the first commercial product in our portfolio of algae-based B2B ingredients," noted Mr. Tuan.
Solasta™ Astaxanthin is manufactured in accordance with industry best practices and guidelines (current Good Manufacturing Practice - GMP) defined by the United States Food and Drug Administration (FDA). Solasta™ meets the strict quality standards set forth in the Astaxanthin Esters Monographs of the Food Chemical Codex and the US Pharmacopeia.


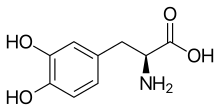



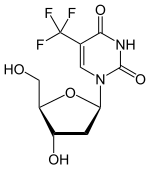 (Trifluridine)
(Trifluridine) 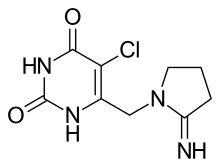 (Tipiracil)
(Tipiracil)





