

Axovant Sciences Ltd. (NYSE: AXON), a leading clinical-stage biopharmaceutical company focused on the treatment of dementia, recently announced the first patients screened in MINDSET, a confirmatory global phase 3 study of Axovant's lead product candidate, RVT-101. Axovant also announced that the company and the U.S. Food and Drug Administration (FDA) have agreed to a Special Protocol Assessment (SPA) supporting this phase 3 program.
MINDSET is an international, multi-center, double blind, placebo-controlled study designed to evaluate the safety, tolerability and efficacy of RVT-101 in patients with mild-to-moderate Alzheimer's disease. The 24-week trial will compare 35-mg, once-daily oral doses of RVT-101 to placebo in approximately 1,150 patients with mild-to-moderate Alzheimer's disease on a stable background of donepezil therapy. The primary efficacy evaluations are the Alzheimer's Disease Assessment Scale – cognitive subscale (ADAS-cog) and the Alzheimer's Disease Cooperative Study – Activities of Daily Living scale (ADCS-ADL), each of which have been used as endpoints to obtain regulatory approval of currently-marketed Alzheimer's disease treatments in the United States and Europe.
The MINDSET trial is designed to confirm the results of a 684-patient international, multi-center, double-blind placebo-controlled study in which patients on a stable background of donepezil therapy receiving 35 mg RVT-101 demonstrated statistically significant improvements on the ADAS-cog and ADCS-ADL as compared to patients receiving donepezil alone.
"I am grateful for the unwavering efforts of the entire development team that has so rapidly advanced RVT-101 into this final stage of the drug development process," said Axovant Chief Development Officer Dr. Lawrence Friedhoff, who is leading the RVT-101 development program and previously led the development program for donepezil (brand name Aricept®), the most widely used Alzheimer's treatment.
"No new compounds have been approved for Alzheimer's disease in over a decade, and physicians are scrambling to do more for their patients," said Dr. Gary Small, President of the American Association for Geriatric Psychiatry. "We need well-tolerated, once-daily oral treatments that provide clinically meaningful benefits. The start of the MINDSET study is an important milestone for the field of Alzheimer's drug development."







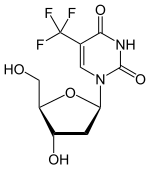 (Trifluridine)
(Trifluridine) 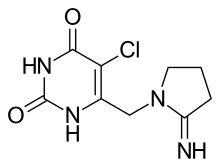 (Tipiracil)
(Tipiracil)



