
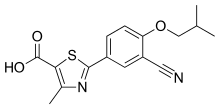
TWi Biotechnology Inc. today announced interim results from the ongoing Phase 2 proof of concept clinical study evaluating AC-201 CR as an oral uricosuric and anti-inflammatory agent for the treatment of hyperuricemia and prevention of gout flares combining with febuxostat, (see structure) a xanthine oxidase inhibitor.
The interim analysis is for the first enrollment cohort (18 per arm completing Week 8), if the number of subjects in the AC-201 CR arm achieving the clinical target of sUA <6.0 mg/dL at Week 8 is at least 2 greater than the number in the placebo arm, then the remaining subjects will be enrolled in the second cohort; if not, enrollment will be stopped. Based on the report, the responders in AC-201 CR arm is at least 2 greater than the number in the placebo arm. In addition to producing the efficacy of AC-201 CR in gout patients, there have been no serious adverse events reported to date and AC-201 CR was generally safe and well tolerated in the study patients. Therefore, TWIB is able to continue to enroll second cohort subjects and complete this clinical trial in the end of 2016.
"We are encouraged to see the potential efficacy of AC-201 CR in the Phase 2 proof-of-concept study results to date," said Dr. Calvin C. Chen, President and CEO of TWIB. "Current evidence of AC-201 CR efficacy and the preliminary results for tolerance support the potential of AC-201 CR as a dual uricosuric and anti-inflammatory agent for the treatment of hyperuricemia and the prevention of gout flares when combining with xanthine oxidase inhibitor. I am very pleased with the interim data as it indicates AC-201 CR may increase the successful rate of the treatment of gout without increase or even decrease the flare rate. The poor compliance of the urate lowering therapy due to frequent gout flares at the initiation and low treatment successful rate offered by existing therapies are the major unmet needs for the management of sUA and gout."











 Temsirolimus
Temsirolimus Ibrutinib
Ibrutinib