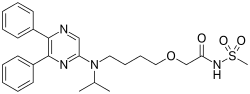
AcelRx Pharmaceuticals, Inc. (Nasdaq: ACRX), a specialty pharmaceutical company focused on the development and commercialization of innovative therapies for the intended treatment of acute pain, reported that the Committee for Medicinal Products for Human Use (CHMP) at the European Medicines Agency (EMA) has confirmed that a Marketing Authorization Application (MAA) for ARX-04 may be submitted in the European Union (EU) under the Agency's centralized procedure. ARX-04 (sufentanil sublingual tablet, 30 mcg) is being developed for the treatment of moderate-to-severe acute pain in adult patients administered by a healthcare professional in a medically supervised setting.

ARX-04 is a non-invasive, fast-onset sufentanil product candidate under investigation.
"We appreciate the CHMP's consideration of ARX-04 for regulatory review under the centralized process, which we expect will make our planned MAA filing with the EMA much more efficient," commented Dr. Pamela Palmer, co-founder and chief medical officer of AcelRx. "The EMA's centralized procedure is typically limited to products the EMA believes constitute significant therapeutic, scientific or technical innovations, characteristics we believe ARX-04 possesses."
For eligible drugs, the centralized procedure permits the submission of a single marketing application to the EMA that, if approved, allows the drug to be marketed in all 27 EU member states, as well as in the three European Economic Area (EEA) countries: Iceland, Liechtenstein and Norway.
ARX-04 is currently being studied in an open-label Phase 3 study (SAP302) in adult patients who present in the emergency room with acute moderate-to-severe pain associated with trauma or injury, and an additional Phase 3 study (SAP303) is planned in postoperative patients with acute moderate-to-severe pain. An earlier Phase 3 trial (SAP301) in patients with acute moderate-to-severe pain following ambulatory abdominal surgery demonstrated that patients administered ARX-04 experienced significantly greater pain relief compared to those receiving placebo, as measured by the time-weighted summed pain intensity difference over the first 12 hours of treatment (SPID12) (p<0.001). AcelRx expects to file marketing applications to both the EMA and the U.S. Food and Drug Administration (FDA) for ARX-04.