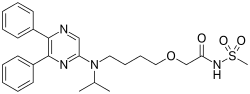
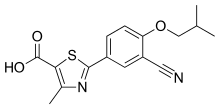
"Bridion provides a new treatment option that may help patients recover sooner from medications used for intubation or ventilation during surgery,” said Sharon Hertz, The U.S. Food and Drug Administration today approved Bridion (sugammadex) injection to reverse the effects of neuromuscular blockade induced by rocuronium bromide and vecuronium bromide, which are used during certain types of surgery in adults.
 M.D., director of the Division of Anesthesia, Analgesia and Addiction Products in the FDA’s Center for Drug Evaluation and Research. “This drug enables medical personnel to reverse the effects of neuromuscular blocking drugs and restore spontaneous breathing after surgery.
M.D., director of the Division of Anesthesia, Analgesia and Addiction Products in the FDA’s Center for Drug Evaluation and Research. “This drug enables medical personnel to reverse the effects of neuromuscular blocking drugs and restore spontaneous breathing after surgery.
Rocuronium bromide and vecuronium bromide are neuromuscular blocking drugs that cause temporary paralysis by interfering with the transmission of nerve impulses to the muscle and are used to paralyze the vocal cords when patients require an artificial airway or breathing tube for surgery, a process called tracheal intubation. They can also be used to prevent patients from moving during surgery while they are receiving general anesthesia. Neuromuscular blocking drugs are also sometimes used to prevent the body from breathing automatically when a patient has to be placed on a ventilator.