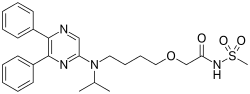
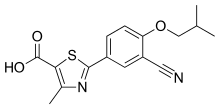
Celecoxib/amlodipine besylate (KIT-302)


itov Pharmaceuticals, an innovative biopharmaceutical company focused on late-stage drug development, announced today that the Phase III, double-blind, placebo-controlled clinical trial for its leading drug candidate, KIT-302, successfully met the primary efficacy endpoint of the trial protocol as approved by the U.S. Food & Drug Administration (FDA). Data from the trial further revealed that KIT-302 was more efficacious at reducing hypertension than the widely used hypertension drug amlodipine besylate. Kitov plans to file its New Drug Application (NDA) for marketing approval of KIT-302 with the FDA in the second half of 2016.
A combination drug, KIT-302, simultaneously treats pain caused by osteoarthritis and treats hypertension, which is a common side effect of stand-alone drugs that treat osteoarthritis pain. KIT-302 is comprised of two FDA approved drugs, celecoxib (Celebrex®) for the treatment of pain caused by osteoarthritis and amlodipine besylate, a drug designed to treat hypertension.
The trial protocol, approved by the FDA through the Special Protocol Assessment process, was designed to quantify the decrease of hypertension in patients receiving KIT-302. The trial was performed in the U.K. in four groups of twenty-six (26) to forty-nine (49) patients, with a total of 152 patients. Each patient was treated over a total period of two weeks. Group One was treated with KIT-302, comprised of celecoxib and amlodipine besylate. Group Two was treated with amlodipine besylate only, one of the components of KIT-302. Group Three was treated with celecoxib only, the other component of KIT-302. Group Four was treated with a double placebo. The trial began in June 2014 and was completed in November 2015.