Sildenafil may be better given to pulmonary arterial hypertension patients in combination with ambrisentan than with bosentan, study findings suggest.
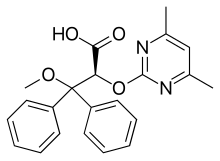 (Ambrisentan)
(Ambrisentan) 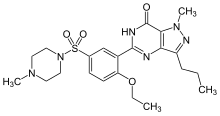
(Sildenafil)
Sildenafil may be better given to pulmonary arterial hypertension (PAH) patients in combination with ambrisentan than with bosentan, study findings suggest.Researcher Keiichi Odagiri (Hamamatsu University School of Medicine, Japan) and team found that patients’ plasma concentration of sildenafil was significantly lower if given with bosentan, compared with ambrisentan.
They say this is because bosentan induces the expression of cytochrome P450 3A4, thus interfering with the pharmacokinetics of sildenafil. Ambrisentan has no such effect, so when the seven study patients, who were all taking sildenafil, were switched from bosentan to ambrisentan, their plasma concentrations of sildenafil were significantly higher.
The patients attained maximum sildenafil concentration in 1.0 hours when they took it with ambrisentan, compared with 0.5 hours with bosentan, and their respective maximum plasma concentrations were 120.2 versus 58.3 ng/mL.And the corresponding areas under the plasma concentration–time curve, representing plasma concentrations across 8 hours, were 396.8 versus 165.8 ng/h per mL.
The team notes that the patients received bosentan 62.5 mg twice daily, rather than the usually recommended 125.0 mg twice daily. They explain that Japanese physicians often use this dose “because of concerns about dose-dependent hepatic toxicity, even though the pharmacokinetics of bosentan and its metabolites are broadly comparable in Japanese and Caucasian individuals.”
Patients’ exercise tolerance improved in line with the higher sildenafil levels achieved with ambrisentan versus bosentan, the researchers report in Clinical and Translational Science.
The median distance achieved in the externally paced 10-metre shuttle walking test was significantly greater during 4–5 weeks of ambrisentan treatment than during the equivalent time on bosentan, at 340 versus 280 metres. The corresponding median 6-minute walking distances were not significantly different, at 503.0 versus 454.0 metres, but peak oxygen consumption and oxygen consumption at anaerobic threshold were significantly greater during the ambrisentan versus bosentan treatment periods.
The researchers caution, however, that they did not use invasive haemodynamic measures to definitively measure treatment response.

 (Elbasvir)
(Elbasvir)  (Grazoprevir)
(Grazoprevir)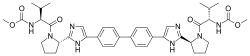
 Tetracaine
Tetracaine