No evidence found linking anti-nausea drug to birth defects: No evidence has been found to link the anti-nausea drug Zofran to an increased risk of birth defects. In fact, women with the condition who took Zofran reported fewer miscarriages and pregnancy terminations and higher live birth rates than women with extreme morning sickness who did not take the drug.

Med-Chemist : "Quintessential Medicinal Chemistry"
Monday, May 9, 2016
Novartis receives FDA Breakthrough Therapy designation for PKC412 (midostaurin)

Novartis announced today that the United States Food and Drug Administration (FDA) has granted Breakthrough Therapy designation to PKC412 (midostaurin). PKC412 (midostaurin) is an investigational treatment for adults with newly-diagnosed AML who are FLT3 mutation-positive, as detected by an FDA-approved test, and who are eligible to receive standard induction and consolidation chemotherapy.
The Breakthrough Therapy designation for PKC412 (midostaurin) is primarily based upon the positive results from the Phase III RATIFY (CALGB 10603) clinical trial. This study was conducted in partnership with the Alliance for Clinical Trials in Oncology and presented during a plenary session at the 57th American Society of Hematology (ASH) Annual Meeting.
Patients who received PKC412 (midostaurin) and standard induction and consolidation chemotherapy experienced a significant improvement in overall survival (OS) (hazard ratio = 0.77, P = 0.0074) compared to those who received standard induction and consolidation chemotherapy alone. The median OS for patients in the PKC412 (midostaurin) treatment group was 74.7 months (95% confidence interval [CI]: 31.7, not attained), versus 25.6 months (95% CI: 18.6, 42.9) for patients in the placebo group. No statistically significant differences were observed in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in the PKC412 (midostaurin) treatment group versus the placebo group. A total of 37 deaths were reported, with no difference in treatment-related deaths observed between groups.
"For more than 25 years, medical developments have been limited for AML patients and the chemotherapy treatment strategy has essentially remained unchanged," said Alessandro Riva, MD, Global Head, Novartis Oncology Development and Medical Affairs. "We look forward to working closely with the FDA to bring PKC412 (midostaurin), the first potential AML targeted therapy, to patients as quickly as possible."
Friday, May 6, 2016
Lenalidomide trials show potential for expanding lymphoma, leukaemia indications

Positive findings from two clinical trials have been published for the immunomodulatory agent lenalidomide in patients with heavily pretreated mantle cell lymphoma, and in adults with T-cell leukaemia-lymphoma or peripheral T-cell lymphoma.
The results of the phase II MCL-002 (SPRINT) study suggest that, compared with an investigator's choice of treatment, lenalidomide 25 mg/day on days 1-21 of a 28-day cycle significantly improved progression-free survival (PFS) in patients with relapsed or refractory mantle cell lymphoma who were ineligible for intensive chemotherapy or stem cell transplantation.
After a median of 15.9 months, the 170 lenalidomide-treated patients had a median PFS of 8.7 months compared with 5.2 months in the 84 patients who were treated with single-agent rituximab, gemcitabine, fludarabine, chlorambucil or cytarabine, giving a hazard ratio (HR) of 0.61.
Tuesday, May 3, 2016
FDA Approves Zepatier (elbasvir and grazoprevir) for Chronic Hepatitis C Genotypes 1 and 4
 (Elbasvir)
(Elbasvir)  (Grazoprevir)
(Grazoprevir)
The U.S. Food and Drug Administration today approved Zepatier (elbasvir and grazoprevir) with or without ribavirin for the treatment of chronic hepatitis C virus (HCV) genotypes 1 and 4 infections in adult patients.
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. Most people infected with HCV have no symptoms of the disease until liver damage becomes apparent, which may take several years. Some people with chronic HCV infection develop cirrhosis over many years, which can lead to complications such as bleeding, jaundice (yellowish eyes or skin), fluid accumulation in the abdomen, infections or liver cancer. According to the Centers for Disease Control and Prevention, approximately 3 million Americans are infected with HCV, of which genotype 1 is the most common and genotype 4 is one of the least common.
“Today’s approval provides another oral treatment option for patients with genotypes 1 and 4 HCV infections without requiring use of interferon,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Monday, May 2, 2016
FDA Approves Expanded Use of Daklinza (daclatasvir) for Additional Challenging-to-treat Patients with Genotype 1 or Genotype 3 Chronic Hepatitis C
In continuation of my update on daclatasvir
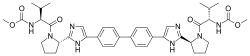
Bristol-Myers Squibb Company (NYSE:BMY) announced today that Daklinza (daclatasvir, 60 mg), an NS5A replication complex inhibitor, has been approved by the U.S. Food and Drug Administration (FDA) in combination with sofosbuvir (with or without ribavirin) in genotypes 1 and 3. The expanded label includes data in three additional challenging-to-treat patient populations: chronic hepatitis C virus (HCV) patients with HIV-1 coinfection, advanced cirrhosis, or post-liver transplant recurrence of HCV. The Daklinza plus sofosbuvir regimen is already available for the treatment of chronic HCV genotype 3, and is currently the only 12-week, once-daily all-oral treatment option for these patients. Sustained virologic response (SVR) rates are reduced in genotype 3 patients with cirrhosis receiving Daklinza and sofosbuvir for 12 weeks without ribavirin. The recommended dosage of Daklinza is 60 mg in combination with sofosbuvir with or without (+/-) ribavirin for 12 weeks.
St. Renatus, LLC Announces FDA Approval of Kovanaze (tetracaine HCl and oxymetazoline HCl) Nasal Spray for Use in Dentistry
St. Renatus, LLC, a privately held company based in Fort Collins, Colorado, announced the U.S. Food and Drug Administration (FDA) approval on June 29, 2016 for its first product, a new dental anesthetic, Kovanaze (tetracaine HCl and oxymetazoline HCl) Nasal Spray. This is the first product that allows for dental anesthesia to be administered through a nasal spray without using a needle.
 Tetracaine
Tetracaine 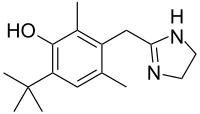 Oxymetazoline
Oxymetazoline
"For more than 100 years, the dental industry has delivered dental anesthesia using a needle injection. Now, through the efforts of a dedicated team, we have developed a revolutionary needle-free method for delivering pulpal anesthesia," said Steve Merrick, St. Renatus' CEO.
Kovanaze is intended for use in dentistry as a topical anesthetic, delivered in the nasal cavity to achieve pulpal (tooth nerve) anesthesia for the restorative treatment of teeth. Like traditional dental injections, this product delivers a local dental anesthetic but without the needle.
Kovanaze is indicated for regional anesthesia when performing a restorative procedure on Teeth 4-13 and A-J in adults and children who weigh 40 kg or more.
Friday, April 29, 2016
FDA Approves Two Supplemental Indications for Harvoni in Chronic Hepatitis C Patients With Advanced Liver Disease
In continuation of my update on Harvoni (ledipasvir/sofosbuvir)
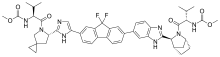
Gilead Sciences, Inc. (NASDAQ: GILD) today announced that the U.S. Food and Drug Administration (FDA) has approved additional indications for Harvoni (ledipasvir/sofosbuvir) for use in chronic hepatitis C patients with advanced liver disease. Harvoni in combination with ribavirin (RBV) for 12 weeks was approved for use in chronic hepatitis C virus (HCV) genotype 1- or 4-infected liver transplant recipients without cirrhosis or with compensated cirrhosis (Child-Pugh A), and for HCV genotype 1-infected patients with decompensated cirrhosis (Child-Pugh B or C), including those who have undergone liver transplantation. Harvoni is now approved for use in a broader range of patient populations, including HCV genotypes 1, 4, 5 and 6, HCV/HIV-1 coinfection, HCV genotype 1 and 4 liver transplant recipients, and genotype 1-infected patients with decompensated cirrhosis..
Thursday, April 28, 2016
Arbor Pharmaceuticals Announces FDA Approval of Cetylev
Arbor Pharmaceuticals announced today that the U.S. Food and Drug Administration (FDA) approved its New Drug Application (NDA) for Cetylev (acetylcysteine effervescent tablets for oral solution).

(acetylcysteine)
Cetylev is an antidote for acetaminophen overdose indicated to prevent or lessen hepatic injury after ingestion of a potentially hepatotoxic quantity of acetaminophen in patients with acute ingestion or from repeated supratherapeutic ingestion. Acetaminophen overdose is the most common poisoning reported to emergency rooms in the United States. According to the American Association of Poison Control Centers there were 73,347 reported acetaminophen exposures in 2014 which resulted in 108 deaths.
Cetylev will be available in ready to use effervescent tablets intended to be mixed with water, resulting in a pleasant lemon-mint tasting solution. Currently, acetylcysteine solution is available in vials intended for inhalation which pharmacists typically mix with a diet cola for ingestion orally. It is also available in an intravenous dosage form which requires that patients be admitted into the hospital.
"For patients with acetaminophen overdose, it is critically important to administer acetylcysteine as quickly as possible to reverse or reduce potential fatal damage to the liver. Emergency rooms, ambulances and pharmacies will have a ready to use dosage form of acetylcysteine that can be administered quickly," said Ed Schutter, President and CEO of Arbor. "Cetylev adds to our growing portfolio of now nineteen different approved prescription products that may help to improve the lives of our patients."
Wednesday, April 27, 2016
Study finds no beneficial effect of cancer medication on Alzheimer's disease

Bexarotene (brand name: Targretin) is an antineoplastic (anti-cancer) agent approved by the U.S.Food and Drug Administration (FDA) (in late 1999) and the European Medicines Agency (EMA) (early 2001) for use as a treatment for cutaneous T cell lymphoma (CTCL). It is a third-generation retinoid.
Study finds no beneficial effect of cancer medication on Alzheimer's disease: The cancer medication bexarotene, aka Targretin, made headlines when researchers reported that it quickly lowered Aβ in the brain, reduced amyloid plaques, and improved learning and memory in mice that mimic salient features of Alzheimer's disease (AD).
Tuesday, April 26, 2016
Impax Receives Approval of Emverm (mebendazole) Chewable Tablets
Impax Laboratories, Inc. (NASDAQ: IPXL) today announced that the United States Food and Drug Administration (FDA) has approved the Company's supplemental new drug application (sNDA) for Emverm (mebendazole) 100 mg chewable tablets.

Impax Receives Approval of Emverm (mebendazole) Chewable Tablets
Monday, April 25, 2016
Allergan Announces FDA Approval of Updated Label for New Dosing Regimen for Dalvance (dalbavancin)

In continuation of my update on Dalbavancin
Allergan plc (NYSE: AGN), a leading global pharmaceutical company, today announced the U.S. Food and Drug Administration (FDA) has approved the company's supplemental new drug application (sNDA) to update the label for Dalvance (dalbavancin) for injection. The expanded label will include a single dose administered as a 30-minute intravenous (IV) infusion of Dalvance for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by designated susceptible Gram-positive bacteria in adults, including infections caused by methicillin-resistant Staphylococcus aureus (MRSA).
Thursday, April 21, 2016
Adzenys XR-ODT (amphetamine) FDA Approval History
Neos Therapeutics, Inc. (Nasdaq:NEOS), a pharmaceutical company with a late‐stage pipeline of innovative extended-release (XR) product candidates for the treatment of attention-deficit/hyperactivity disorder (ADHD), today announced that the U.S. Food and Drug Administration (FDA) approved Adzenys XR-ODT for the treatment of ADHD in patients six years and older. With this approval, Adzenys XR-ODT is the first and only extended-release orally disintegrating tablet (ODT) for the treatment of ADHD.
“The novel features of an extended-release orally disintegrating tablet, which is dosed once daily and disintegrates in the mouth, make Adzenys XR-ODT attractive for use in both children (six years and older) and adults,” said Dr. Alice Mao, Medical Director, Memorial Park Psychiatry in Houston, TX.
Adzenys XR-ODT was approved by the FDA via the 505(b)(2) regulatory pathway. The clinical program demonstrated that Adzenys XR-ODT is bioequivalent to a previously approved mixed amphetamine salts extended-release capsule (Adderall XR1), one of the most commonly prescribed medications for the treatment of ADHD. Adzenys XR-ODT will be available in six dosage strengths, equivalent to the Adderall XR1 dosage strengths, thus allowing healthcare providers to individualize the dose.
FDA Approves Halaven (eribulin mesylate) for the Treatment of Liposarcoma

The U.S. Food and Drug Administration today approved Halaven (eribulin mesylate), a type of chemotherapy, for the treatment of liposarcoma (a specific type of soft tissue sarcoma) that cannot be removed by surgery (unresectable) or is advanced (metastatic). This treatment is approved for patients who received prior chemotherapy that contained an anthracycline drug.
“Halaven is the first drug approved for patients with liposarcoma that has demonstrated an improvement in survival time,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The clinical trial data the FDA reviewed indicates that Halaven increased overall survival by approximately seven months, offering patients a clinically meaningful drug.”
Wednesday, April 20, 2016
FDA Approves Onzetra Xsail (sumatriptan nasal powder) for the Acute Treatment of Migraine

Sumatriptan is a medication used for the treatment of migraine headaches.It is a synthetic drug belonging to the triptan class. Structurally, it is an analog of the naturally occurring neuro-active alkaloidsdimethyltryptamine (DMT), bufotenine, and 5-methoxy-dimethyltryptamine, with an N-methyl sulfonamidomethyl- group at position C-5 on theindole ring.
Sumatriptan is produced and marketed by various drug manufacturers with many different trade names such as Imitrex, Imigran, and Treximetas a combination product with naproxen.
Avanir Pharmaceuticals, Inc. today announced that the U.S. Food and Drug Administration (FDA) has approved Onzetra Xsail (sumatriptan nasal powder), formerly known as AVP-825, for the acute treatment of migraine with or without aura in adults. Onzetra Xsail is an intranasal medication delivery system consisting of a low-dose (22mg) of sumatriptan powder that is delivered utilizing the novel Xsail™ Breath Powered Delivery Device. Onzetra Xsail is a fast-acting dry powder formulation of sumatriptan, the most commonly prescribed migraine medication.
“While there are many acute migraine treatment options available, more than 70% of patients are not fully satisfied with their current migraine treatment. Given this high dissatisfaction, there remains an unmet need to provide patients with fast-acting, well tolerated therapies that deliver consistent relief,” said Stewart Tepper, M.D., professor of neurology at the Geisel School of Medicine at Dartmouth.
“Onzetra Xsail provides a new and much needed treatment option for what can be a debilitating condition for millions of people,” said Roger K. Cady, M.D., director of the Headache Care Center and associate executive chairman of the National Headache Foundation. “The Xsail Breath Powered Delivery Device allows the medication to be deposited deep into the nose, an area that is rich with blood vessels. By delivering the medication here, Onzetra Xsail provides targeted and efficient delivery with the potential for fast, consistent relief, while also limiting the amount of medicine that goes down the back of the throat.”
“We are excited about this major milestone for Avanir as the approval of Onzetra Xsail represents our second approved product. We expect to make Onzetra Xsail available to patients in the coming months,” said Rohan Palekar, president and CEO of Avanir. “We continue to be focused on building a leading CNS biopharmaceutical company and the addition of Onzetra Xsail to our product portfolio takes us closer to achieving that goal.”
Tuesday, April 19, 2016
FDA Approves Takeda's Dexilant SoluTab (dexlansoprazole)
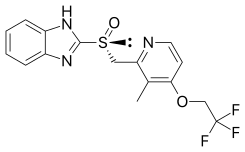
Dexlansoprazole (INN, trade names Kapidex, Dexilant) is a proton pump inhibitor that is marketed by Takeda Pharmaceuticals for the treatment of erosive esophagitis and gastro-oesophageal reflux disease...
Takeda Pharmaceuticals U.S.A., Inc., (Takeda) (TSE: 4502) today announced that the United States (U.S.) Food and Drug Administration (FDA) approved Dexilant SoluTab delayed-release orally disintegrating tablets, a new formulation of dexlansoprazole that can be taken by allowing the tablet to melt in the patient's mouth. Dexilant SoluTab is a proton pump inhibitor (PPI) indicated for the treatment of heartburn associated with symptomatic non-erosive gastroesophageal reflux disease (GERD) and the maintenance of healed erosive esophagitis (EE) and relief of heartburn in adults 18 years and older. Dexilant SoluTab is a PPI with dual delayed release (DDR) technology that is designed to provide two separate releases of medication.
Subscribe to:
Posts (Atom)