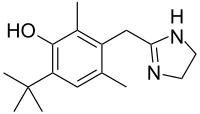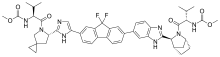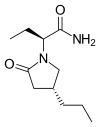
The U.S. Food and Drug Administration yesterday approved Briviact (brivaracetam) as an add-on treatment to other medications to treat partial onset seizures in patients age 16 years and older with epilepsy.
Epilepsy is a brain disorder that causes people to have recurring seizures. A seizure is an episode, usually of relatively short duration, of abnormal brain activity. Seizures can cause a variety of symptoms, including uncontrolled movements or spasms, abnormal thinking and behavior, and abnormal sensations. Muscle spasms can be violent, and loss of consciousness can occur. Seizures occur when clusters of nerve cells (neurons) in the brain undergo uncontrolled activation. A partial onset seizure begins in a limited area of the brain.
“Patients can have different responses to the various seizure medicines that are available,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “With the approval of Briviact, I am pleased that patients with epilepsy have a new treatment option.”
FDA Approves Briviact (brivaracetam) to Treat Partial Onset Seizures

 (
( (Donepezil)
(Donepezil) 

 (Elbasvir)
(Elbasvir)  (Grazoprevir)
(Grazoprevir)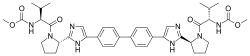
 Tetracaine
Tetracaine