
Gilead Sciences, Inc announced that the U.S. Food and Drug Administration (FDA) has approved Odefsey (emtricitabine 200 mg/rilpivirine 25 mg/tenofovir alafenamide 25 mg or R/F/TAF) for the treatment of HIV-1 infection in certain patients. Emtricitabine and tenofovir alafenamide are from Gilead Sciences and rilpivirine is from Janssen Sciences Ireland UC, one of the Janssen Pharmaceutical Companies of Johnson & Johnson (Janssen). Odefsey is Gilead’s second TAF-based regimen to receive FDA approval and represents the smallest pill of any single tablet regimen for the treatment of HIV.
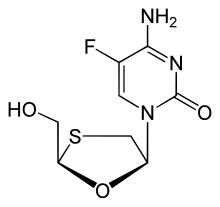 Emtricitabine
Emtricitabine  Rilpivirine
Rilpivirine  Tenofovir alafenamide
Tenofovir alafenamide
Odefsey is indicated as a complete regimen for the treatment of HIV-1 infection in patients 12 years of age and older who have no antiretroviral treatment history and HIV-1 RNA levels less than or equal to 100,000 copies per mL. Odefsey is also indicated as replacement for a stable antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) for at least six months with no history of treatment failure and no known substitutions associated with resistance to the individual components of Odefsey. No dosage adjustment of Odefsey is required in patients with estimated creatinine clearance greater than or equal to 30 mL per minute.
Odefsey has a boxed warning in its product label regarding the risks of lactic acidosis/severe hepatomegaly with steatosis, and post treatment acute exacerbation of hepatitis B.
TAF is a novel targeted prodrug of tenofovir that has demonstrated high antiviral efficacy similar to and at a dose less than one-tenth that of Gilead’s Viread (tenofovir disoproxil fumarate, TDF). TAF has also demonstrated improvement in surrogate laboratory markers of renal and bone safety as compared to TDF in clinical trials in combination with other antiretroviral agents. Data show that because TAF enters cells, including HIV-infected cells, more efficiently than TDF, it can be given at a much lower dose and there is 90 percent less tenofovir in the bloodstream.



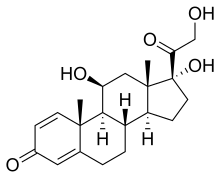 (Prednisole)
(Prednisole) (Indomethacin)
(Indomethacin)
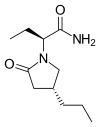
 (
( (Donepezil)
(Donepezil) 