
Allergan plc, a leading global pharmaceutical company, today announced that the Company has received approval from the U.S. Food and Drug Administration (FDA) to market Aczone (dapsone) Gel, 7.5%, a new prescription topical treatment for acne in patients 12 years of age and older. Aczone Gel, 7.5% delivers proven efficacy to treat both inflammatory and non-inflammatory acne, with a new concentration of dapsone in just once-a-day application.
 (Dapsone)
(Dapsone)
"For the Aczone Gel, 7.5% pivotal trials, we studied 4,340 acne patients, demonstrating efficacy and tolerability. The new FDA product approval also offers just once-daily dosing and a new pump delivery system," said David Nicholson, Executive Vice President and President of Global R&D Brands at Allergan. "As part of Allergan's commitment to the medical dermatology space, we have truly raised the bar for ourselves in efforts to offer an effective acne product to address physician and patient needs."
In clinical trials of Aczone Gel, 7.5%, safety and efficacy were assessed in two identically designed, randomized, multi-centered, double-blind, vehicle-controlled 12-week studies. A total of 4,340 acne patients were randomized to receive either Aczone Gel, 7.5% (n=2162) or vehicle (n=2178). The majority of patients (99%; n=4339) had moderate acne, with a baseline score of 3 on the Global Acne Assessment Score (GAAS). Aczone Gel, 7.5% was approved based on co-primary endpoints of the GAAS and lesion counts (20 to 50 inflammatory and 30 to 100 non-inflammatory lesions at baseline). At week 12, inflammatory lesions were reduced by 15.8 lesions (54.6%; n=2162) vs 13.9 lesions with vehicle (48.1%; n=2178), and non-inflammatory lesions were reduced by 20.7 lesions (45.1%) vs 18.0 lesions with vehicle (39.4%).2 The GAAS success rate in patients was 29.8% (n=2162) vs 21.1% with vehicle (n=2178).
In addition to efficacy, Aczone Gel, 7.5% has a proven tolerability profile. Out of 2161 patients who used Aczone Gel, 7.5%, 1.1% experienced mild application-site dryness vs 1.0% with vehicle (n=2175), and 0.9% experienced pruritus vs 0.5% with vehicle.
 Palbociclib
Palbociclib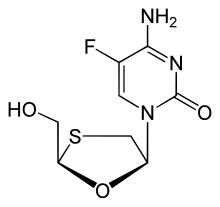 Emtricitabine
Emtricitabine  Rilpivirine
Rilpivirine Tenofovir alafenamide
Tenofovir alafenamide


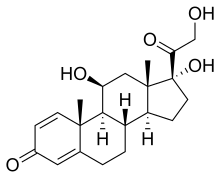 (Prednisole)
(Prednisole) (Indomethacin)
(Indomethacin)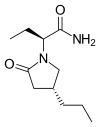
 (
( (Donepezil)
(Donepezil)