
In continuation of my update on Melatonin
Melatonin, a hormone produced in the human brain, appears to suppress the growth of breast cancer tumors.
Researchers at Michigan State University published this finding in the current issue ofGenes and Cancer. While treatments based on this key discovery are still years away, the results give scientists a key foundation on which to build future research.

"You can watch bears in the zoo, but you only understand bear behavior by seeing them in the wild," said David Arnosti, MSU biochemistry professor, director of MSU's Gene Expression in Development and Disease Initiative and co-author of the study. "Similarly, understanding the expression of genes in their natural environment reveals how they interact in disease settings. That's what is so special about this work."
The brain manufactures melatonin only at night to regulate sleep cycles. Epidemiologists and experimentalists have speculated that the lack of melatonin, due in part to our sleep-deprived modern society, put women at higher risk for breast cancer. The latest MSU study showed that melatonin suppresses the growth of breast cancer stem cells, providing scientific proof to support the growing body of anecdotal evidence on sleep deprivation.
The research team was led by Juliana Lopes, a visiting researcher from Sao Paolo, Brazil. Before the team could test its theory, the scientists had to grow tumors from stem cells, known as "mammospheres," a method perfected in the laboratory of James Trosko at MSU.
The growth of these mammospheres was enhanced with chemicals known to fuel tumor growth, namely, the natural hormone estrogen, and estrogen-like chemical Bisphenol A, or BPA, found in many types of plastic food packages.
Melatonin treatment significantly decreased the number and size of mammospheres when compared with the control group. Furthermore, when the cells were stimulated by estrogen or BPA and treated with melatonin at the same time, there was a greater reduction in the number and size of mammospheres.
"This work establishes the principal by which cancer stem cell growth may be regulated by natural hormones, and provides an important new technique to screen chemicals for cancer-promoting effects, as well as identify potential new drugs for use in the clinic," Trosko said.




 (Dapsone)
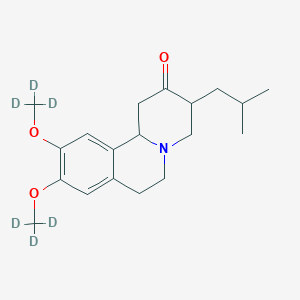
(Dapsone) Palbociclib
Palbociclib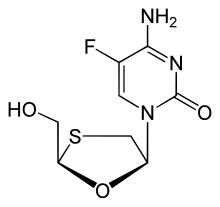 Emtricitabine
Emtricitabine  Rilpivirine
Rilpivirine Tenofovir alafenamide
Tenofovir alafenamide
