
Scientists at Nanyang Technological University, Singapore (NTU Singapore) have discovered that antibiotics can continue to be effective if bacteria's cell-to-cell communication and ability to latch on to each other are disrupted.
This research breakthrough is a major step forward in tackling the growing concern of antibiotic resistance, opening up new treatment options for doctors to help patients fight against chronic and persistent bacterial infections.
The study, led by Assistant Professor Yang Liang from the Singapore Centre for Environmental Life Sciences (SCELSE) at NTU, found that a community of bacteria, known as biofilm, can put up a strong line of defence to resist antibiotics. The NTU team has successfully demonstrated how biofilms can be disrupted to let antibiotics continue their good work.
The research was published recently in Nature Communications, a prestigious academic journal by the Nature Publishing Group.
"Many types of bacteria that used to be easily killed by antibiotics have started to develop antibiotic resistance or tolerance, either through acquiring the antibiotic resistant genes or by forming biofilms," said Asst Prof Yang, who also teaches at NTU's School of Biological Sciences.
"The US Center for Disease Control estimates that over 60 per cent of all bacterial infections are related to biofilms. Our study has shown that by disrupting the cell-to-cell communication between bacteria and their ability to latch on to each other, we can compromise the biofilms, leaving the bacteria vulnerable and easily killed by antibiotics."
Bacterial resistance to antibiotics is rapidly growing world-wide and this puts at risk the ability to treat common infections in the community and hospitals.
The World Health Organisation states on its factsheet on Antimicrobial resistance that "without urgent, coordinated action, the world is heading towards a post-antibiotic era, in which common infections and minor injuries, which have been treatable for decades, can once again kill".
Associate Professor Kevin Pethe, an expert in antibiotic development and infectious diseases from NTU's Lee Kong Chian School of Medicine, said that this discovery may yield new treatment options that doctors can employ against chronic and persistent bacterial infections.
"Being able to disable biofilms and its protective benefits for the bacteria is a big step towards tackling the growing concern of antibiotic resistance," said Assoc Prof Pethe.
"While the scientific community is developing new types of antibiotics and antimicrobial treatments, this discovery may help to buy time by improving the effectiveness of older drugs."

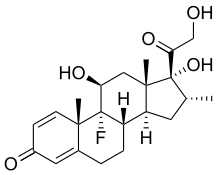




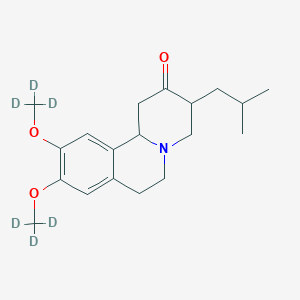

 (Dapsone)
(Dapsone) Palbociclib
Palbociclib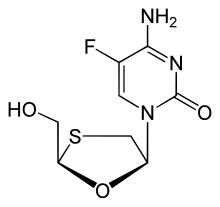 Emtricitabine
Emtricitabine  Rilpivirine
Rilpivirine Tenofovir alafenamide
Tenofovir alafenamide