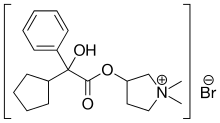
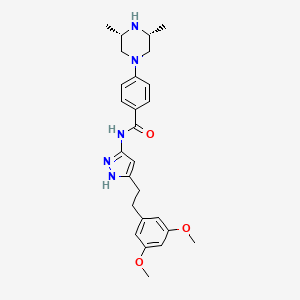
In continuation of my update on Lenvatinib
 Lenvatinib
Lenvatinib
Eisai Inc. announced today that the U.S. Food and Drug Administration (FDA) approved Lenvima (lenvatinib), the company's multiple receptor tyrosine kinase inhibitor, in combination with everolimus for the treatment of patients with advanced renal cell carcinoma(aRCC) who were previously treated with an anti-angiogenic therapy. This approval was based on the impressive results of the registration study (Study 205), in which the once daily combination of 18 mg Lenvima and 5 mg everolimus demonstrated a substantial improvement in progression-free survival (PFS), powerful objective response rate (ORR) and clinically meaningful overall survival (OS) when compared with everolimus alone, a standard of care for patients with aRCC who have received prior anti-angiogenic therapy.
"Lenvatinib plus everolimus is the first and only FDA-approved regimen that successfully combines treatments that employ tyrosine kinase and mTOR inhibition, the primary targets of advanced RCC treatment for the past decade," said Robert Motzer, M.D., Memorial Sloan Kettering Cancer Center, New York, and the principal investigator of the study. "This combination regimen led to enhanced efficacy and helped patients with advanced RCC live longer without disease progression or death than those treated with everolimus alone. These noteworthy findings advance the treatment paradigm for this patient population."
Lenvima was granted Breakthrough Therapy designation by the FDA for this indication, and the application received Priority Review, which is assigned to drugs the FDA believes have the potential to provide a significant improvement in the treatment of a serious condition.
In Study 205, a Phase 2 trial, Lenvima and everolimus (LEN+EVE) resulted in a median PFS nearly three times that of everolimus alone. The median PFS, or the length of time from randomization until disease progression or death, in patients treated with the combination (n=51) was 14.6 months (95% CI: 5.9–20.1) compared with 5.5 months (95% CI: 3.5–7.1) for those treated with everolimus alone (n=50) (HR 0.37; 95% CI: 0.22–0.62). The combination regimen resulted in a 63% reduction in the risk of disease progression or death compared with everolimus alone. The treatment effect of the combination on PFS was supported by a retrospective independent review.
The objective response rate was 37% (95% CI: 24–52) in patients treated with the combination regimen (35% partial response + 2% complete response) compared to 6% (all partial response, 95% CI: 1–17) in patients treated with everolimus alone.
The patients who received LEN+EVE experienced a 10.1-month increase in median OS compared with those who received everolimus monotherapy (25.5 months [95% CI: 16.4–32.1] versus 15.4 months [95% CI: 11.8–20.6]; HR 0.67; 95% CI: 0.42–1.08). This OS analysis was conducted when 63% of deaths had occurred in the combination arm and 74% of deaths had occurred in the everolimus arm.
The safety of this combination regimen was also examined in Study 205. Serious risks from treatment with the combination of Lenvima and everolimus may include hypertension, cardiac dysfunction, arterial thromboembolic events, hepatotoxicity, proteinuria, diarrhea, renal failure and impairment, gastrointestinal perforation and fistula formation, QT interval prolongation, hypocalcemia, reversible posterior leukoencephalopathy syndrome, hemorrhagic events, impairment of thyroid-stimulating hormone suppression/thyroid dysfunction, and embryofetal toxicity. The most common adverse reactions observed in study patients treated with Lenvima and everolimus (greater than 30%) were, in order of decreasing frequency, diarrhea, fatigue, arthralgia/myalgia, decreased appetite, vomiting, nausea, stomatitis/oral inflammation, hypertension, peripheral edema, cough, abdominal pain, dyspnea, rash, weight decreased, hemorrhagic events and proteinuria. The most common serious adverse reactions (greater than or equal to 5%) were renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%) and dyspnea (5%). Adverse reactions led to dose reductions or interruption in 89% of patients receiving Lenvima and everolimus and 54% in patients receiving everolimus. The most common adverse reactions (greater than or equal to 5%) resulting in dose reductions in patients treated with Lenvima and everolimus were diarrhea (21%), fatigue (8%), thrombocytopenia (6%), vomiting (6%), nausea (5%) and proteinuria (5%).
Treating physicians are likely to be familiar with many of the adverse reactions observed for this combination regimen based on their prior experience with these types of drugs. Prescribers may be able to manage certain adverse reactions (such as nausea, vomiting, diarrhea and hypertension) with a proactive plan that includes concomitant medications and/or dose reductions, interruptions and/or discontinuations.
"Rates of renal cell carcinoma have been on the rise over the past several decades, and unfortunately, advanced RCC remains an incurable disease. Since the VEGF pathway is known to be involved in the growth of renal cell tumors, it is important to have a diverse offering of therapeutic options, including treatments that continue to target VEGF inhibition," said Sumanta Kumar Pal, M.D., Assistant Professor, Department of Medical Oncology & Therapeutics Research and Co-Director, Kidney Cancer Program at City of Hope in Duarte, Calif. "The combination regimen of lenvatinib and everolimus provides a new treatment for patients with advanced RCC whose disease continues to progress despite prior treatment with an anti-angiogenic therapy."
Lenvima was first approved in the U.S. on February 13, 2015, for patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (DTC).
"By bringing this breakthrough treatment to patients with advanced RCC, Eisai now offers an efficacious option in a second difficult-to-treat tumor type, just 15 months after its initial approval, and we look forward to continued exploration of LENVIMA in additional malignancies," said Alton Kremer, M.D., Ph.D., Chief Clinical Officer and Chief Medical Officer, Oncology Business Group at Eisai. "This also marks the second time in four months that one of Eisai's oncology treatments has been granted a new indication following Priority Review from the FDA. These milestones, as well as the ongoing development of innovative agents in our pipeline, underscore our steadfast commitment to Eisai's human health care (hhc) mission of identifying and addressing the unmet needs of people living with cancer."
About Study 205
Study 205, the Phase 2 study, was a multicenter, randomized trial in patients (n=153) with unresectable advanced or metastatic RCC who were previously treated with an anti-angiogenic therapy and randomized 1:1:1 to receive a combination of 18 mg LENVIMA plus 5 mg everolimus once a day, LENVIMA only (24 mg once a day) or everolimus only (10 mg once a day) administered orally in continuous 28-day cycles until disease progression or unacceptable toxicity. The primary efficacy endpoint of this study was investigator-assessed PFS. Other endpoints of the study included ORR, OS and safety.
The results of this study were published online in The Lancet Oncology in October 2015, following an oral presentation at the 2015 American Society of Clinical Oncology (ASCO) Annual Meeting.