In continuation of my update on Apixaban
The superiority of apixaban over warfarin in patients with atrial fibrillation is maintained in those taking multiple medications, shows further analysis of the ARISTOTLE trial.
The researchers found superior efficacy of apixaban against the primary thromboembolic endpoint (stroke or systemic embolism) regardless of the number of drugs patients were taking.
By contrast, the advantage of apixaban over warfarin in terms of major bleeding tended to decline in line with the number of drugs patients were taking. The absolute rate reduction per 100 patient-years with apixaban versus warfarin was 1.28 for patients taking up to five medications, falling to 0.82 and 0.66 for those taking six to eight and more than nine drugs, respectively.
"Importantly, the risk reduction of intracranial bleeding did not diminish with an increasing number of concomitant drugs", write the researchers in The BMJ.
"Therefore, the fact that the relative benefit of apixaban over warfarin appears to diminish across groups is due to other types of major bleeding."
They give the example of major gastrointestinal bleeding, which was significantly reduced with apixaban versus warfarin in patients taking up to five drugs, but not in those taking nine or more drugs.
Polypharmacy was common among the 18,201 ARISTOTLE participants, with 76.5% taking at least five concomitant drugs. Patients' average age rose in line with the number of drugs used, as did their stroke and bleeding risk. Patients taking more drugs also had more cardiovascular comorbidities, and also more neurological, renal, endocrine, musculoskeletal, pulmonary, gastrointestinal and haematological comorbidities.
Rates of the primary thromboembolic and bleeding endpoints rose with the number of drugs taken for patients in the apixaban group as well as those in the warfarin group.
Jeroen Jaspers Focks (Radboud University Nijmegen Medical Centre, the Netherlands) and study co-authors stress that "this increased risk of adverse outcomes should be placed in the context of the association between the number of drug treatments and comorbidities present at baseline, indicating a more frail status of patients with polypharmacy."
The researchers suggest that adjusting for these differences would abolish the relationship between the number of drugs used and safety outcomes, but add that the purpose of the study was to use polypharmacy as a marker of patient frailty.
Moreover, increasing frailty did not significantly influence the efficacy of apixaban against stroke or systemic embolism, with the relative risk versus warfarin being 14% among those taking up to five drugs and 24% in those taking more.

 carfilzomib
carfilzomib
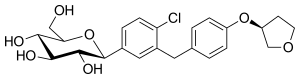 empagliflozin
empagliflozin  In continuation of my update on
In continuation of my update on 
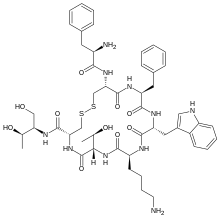
 Solithromycin
Solithromycin
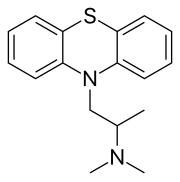 promethazine
promethazine