
In continuation of my update on Pioglitazone

Researchers have found that an existing diabetes drug can be used to halt progression of another disease that is a leading cause of liver transplants.
A three-year clinical trial led by University of Florida Health researcher Kenneth Cusi, M.D., found that the drug pioglitazone is safe and effective in certain patients who have nonalcoholic steatohepatitis, or NASH, a chronic liver disease caused by a buildup of fat. The findings are published today (June 20) in the journal Annals of Internal Medicine.
NASH is often known as "silent" liver disease and affects 10 to 20 percent of the population and perhaps as many as one-third of all patients with adult-onset diabetes in the United States, according to recent studies. Left unchecked, NASH can cause chronic inflammation that leads to liver cancer or cirrhosis. NASH is now the second-leading cause of liver transplants and the numbers continue to grow each year, said Cusi, chief of the division of endocrinology, diabetes and metabolism in the UF College of Medicine's department of medicine.
Early diagnosis and treatment of NASH is crucial for those who are at greatest risk for the disease, usually obese patients who also have prediabetes or Type 2 diabetes. But until now, Cusi said, there was little urgency to diagnose NASH because there were no available medications.
The research group's single-center clinical trial involving 101 NASH patients with prediabetes or Type 2 diabetes found that pioglitazone reduced fatty liver disease activity in 58 percent of participants. In just more than half the participants -- 51 percent -- the disease was reduced enough that it was no longer considered a threat to the liver.
"The exciting thing is that there is a generic drug that already prevents the onset of Type 2 diabetes and cardiovascular disease in recent studies. Now, it can reduce disease from excess liver fat accumulation and liver inflammation, and halt fibrosis that leads to cirrhosis. This will have a lot of long-term benefits for many people with a medication that will be very affordable and is already being used to treat Type 2 diabetes," Cusi said.
The study also has implications for people with prediabetes and NASH because fatty liver disease is a risk factor for Type 2 diabetes even in those who aren't obese, researchers said.
Federal regulators approved Actos (pioglitazone) in 2000 and a generic version of the drug in 2012 to improve blood glucose control in adults with Type 2 diabetes. Still, pioglitazone's use against liver disease will require a larger, multicenter clinical trial that could take seven years or more in addition to U.S. Food and Drug Administration approval. A multicenter trial would allow researchers to learn more about the drug's long-term benefits for liver issues and determine why some participants respond better than others to the medication, Cusi said.
Researchers aren't entirely certain about how pioglitazone works against liver disease. Patients with NASH are insulin-resistant, meaning their body does not respond normally to their own insulin. This defect promotes fat accumulation and inflammation in the liver. The researchers believe the medication makes molecular improvements in the liver and other tissues such as fat. That helps the body's response to insulin, making it insulin-sensitive again and restoring normal metabolism.
Despite the recent trial's relatively small size, Cusi noted that it's the largest single-center study and the first long-term study examining the drug as treatment for people who have NASH along with prediabetes or Type 2 diabetes. It is also the longest NASH-related study with any drug and had the greatest treatment effect on NASH compared with other approaches, he said.
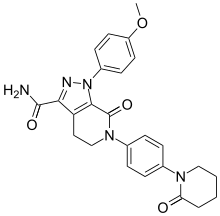

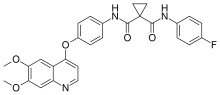
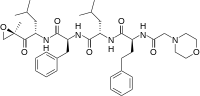 carfilzomib
carfilzomib
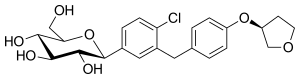 empagliflozin
empagliflozin  In continuation of my update on
In continuation of my update on 
 Solithromycin
Solithromycin
 promethazine
promethazine