A drug that spurs cancer cells to self-destruct has been cleared for use in a clinical trial of patients with anaplastic astrocytoma, a rare malignant brain tumor, and glioblastoma multiforme, an aggressive late-stage cancer of the brain. This phase Ib trial will determine if the experimental drug PAC-1 can be used safely in combination with a standard brain-cancer chemotherapy drug, temozolomide.
The trial is approved for patients who have seen their cancer progress after first-line therapy. This is an extension of an ongoing human phase I clinical trial of PAC-1 alone in patients with various late-stage cancers. Phase I trials are designed to test the safety of new drugs in human patients.
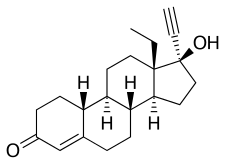
PAC-1 is unusual in that it is able to cross the blood-brain barrier, a formidable obstacle to most anti-cancer drugs. The drug targets procaspase-3, an enzyme that is overexpressed in many cancer cells, said University of Illinois chemistry professor Paul Hergenrother, who discovered PAC-1's anti-cancer effects more than a decade ago. After tests in human cell lines and rodents proved promising, Hergenrother and veterinary oncologist Dr. Timothy Fan, a professor of veterinary clinical medicine at Illinois, tested PAC-1 in pet dogs with a variety of naturally occurring cancers.
"Most cancers have elevated levels of procaspase-3," Hergenrother said. "When it is turned on, procaspase-3 kills cells."
Cancer cells override this normal cell-recycling pathway, however, he said.
"PAC-1 restores the activation of procaspase-3 and, because this enzyme is elevated in cancer cells, targets cancer cells over noncancerous cells," he said.
PAC-1 has been evaluated in pet dogs with naturally occurring osteosarcoma, lymphoma and, most recently, glioma - a brain cancer similar to glioblastoma in humans. One 2016 study found that the combination of PAC-1 with doxorubicin, a chemotherapeutic agent that also is used in humans, saw tumor reductions in four of four dogs with lymphoma and in three of six dogs with osteosarcoma. The trials in dogs continue and, so far, have found PAC-1 to be safe, with few observable side effects apart from occasional gastrointestinal distress. The researchers report their latest findings in rodents and in dogs with brain cancer in the journal Oncotarget.
Dogs with certain naturally occurring cancers may be better than other animal models of human cancers because mice and rats used in many cancer drug-testing models must be implanted with human cancer cells to mimic specific types of tumors, Fan said.
"This requires that the rodents be immunocompromised to mitigate rejection of human cells," he said. "As such, most rodent tumor models do not faithfully recapitulate the tumor microenvironment - in particular, the body's immune surveillance of the tumor.
"Rodent models are limited, but they are still useful," Fan said.
Certain cancers in dogs are genetically similar to those in humans and respond to the same medications. Dogs also are more similar in size to humans, and so can be better models to evaluate how well drug agents perform on larger tumor masses.
"I look at pets with spontaneous tumors as being complementary to rodent models and recognize that not all discoveries in pet dogs will necessarily translate similarly to people," Fan said.
The ongoing clinical trial of PAC-1 in human patients with late-stage solid tumors and lymphoma has shown that the drug is well-tolerated at tested doses up to 450 milligrams per day, said medical oncologist Dr. Arkadiusz Dudek, who chairs an advisory board for Vanquish Oncology, which is funding the clinical trials.
The extension of the phase I trial to brain-cancer patients will begin with a PAC-1 dose of 375 mg per day and will increase the dose incrementally to test its safety in combination with the standard brain-cancer chemotherapy agent, temozolomide, he said.
So far, the clinical trials of PAC-1 alone have seen no significant side effects in humans. None of the human patients in the first five dose levels of the single-agent trial has dropped out as a result of side effects, the researchers report. The team cannot report on clinical outcomes in a phase I clinical trial, since such trials are designed to measure safety, not efficacy.
Surgery is a first-line therapy for anaplastic astrocytoma, followed by treatment with temozolomide, a chemotherapy drug that is one of the few effective treatments for brain cancer, Dudek said. Humans with glioblastoma multiforme usually undergo surgery to remove as much of the cancerous tissue as possible, followed by radiation and oral treatment with temozolomide.
It is almost impossible to find and remove all glioblastoma cancer cells in surgery, however, Dudek said.
"Glioblastoma multiforme has this feature of spreading silently along the blood vessels inside the brain," he said. "That's a reason why most patients will unfortunately have disease coming back later on after surgery and radiation."
The median survival time for human patients with glioblastoma undergoing the standard treatment is about 15 months.
The three dogs in the glioma trial received daily oral doses of PAC-1 in combination with temozolomide and "curative-intent" radiation.
Temozolomide is normally too expensive to use in canine patients, Fan said. The dogs tolerated the combination treatment very well and responded well to the therapy, he said.
"All three dogs had, at the very least, what we call a partial response, which means more than a 30 percent reduction in the tumor," he said. "And one of the dogs had a complete response, as identified with serial MRI scans, with a 100 percent reduction in the tumor mass 84 days after combination therapy."
Fan said a much larger study in dogs would be needed to determine whether the therapeutic effects were consistent and reproducible, and to quantify how much PAC-1 contributed to the positive results.
Vanquish Oncology, a drug-development startup company Hergenrother helped found in 2011, has licensed the technology from the University of Illinois and is focused on moving PAC-1 into the clinic. As with any investigational agent, determining the true safety and efficacy profile of PAC-1 will take several years of human clinical trials.
https://news.illinois.edu/blog/view/6367/583399





 and
and