
Helsinn, a Swiss pharmaceutical group focused on building quality cancer care products, today announces that the U.S. Food and Drug Administration (FDA) has approved the intravenous formulation of Akynzeo (NEPA, a fixed antiemetic combination of fosnetupitant, 235mg, and palonosetron, 0.25mg) as an alternative treatment option for patients experiencing CINV.
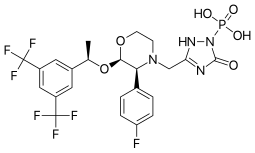
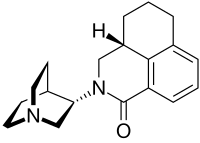
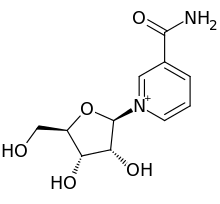
The FDA has approved Akynzeo IV (formulation of fosnetupitant first structure and palonosetron second structure) in combination with dexamethasone in adults for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic cancer chemotherapy. Akynzeo for injection has not been studied for the prevention of nausea and vomiting associated with anthracycline plus cyclophosphamide chemotherapy.
Oral Akynzeo was previously approved by the FDA as a fixed combination oral agent in 2014 for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of cancer chemotherapy, including, but not limited to, highly emetogenic chemotherapy. Akynzeo is an oral fixed combination of palonosetron and netupitant: palonosetron prevents nausea and vomiting during the acute phase and netupitant prevents nausea and vomiting during both the acute and delayed phase after cancer chemotherapy.
The bioequivalence of the IV with the oral formulation of netupitant was demonstrated and the safety of IV NEPA was established through a repeated dose safety study in cancer patients to potentially uncover adverse drug reactions that may appear during subsequent clinical practice. No anaphylactic and injection site reactions related to IV NEPA were reported in this study.
Currently a repeated dose safety study is ongoing in patients receiving anthracycline plus cyclophosphamide to further establish the safety profile in this setting.
The prevention of CINV has been refined in treatment guidelines over the past several decades. Currently the combination treatment of antiemetic medicines with different mechanisms of actions are recommended for the prevention of CINV.
The approval of Akynzeo in IV formulation will offer to US patients and healthcare providers an alternative route of administration of the only fixed antiemetic combination targeting two distinct CINV pathways in a single dose.
Riccardo Braglia, Helsinn Group Vice Chairman and CEO, commented: “The approval of the intravenous formulation of Akynzeo paves the way to bring this important therapeutic option to more patients in a new formulation, and we are delighted that we are now able to push ahead with launching this product in the United States in May 2018”

 Ivacafto
Ivacafto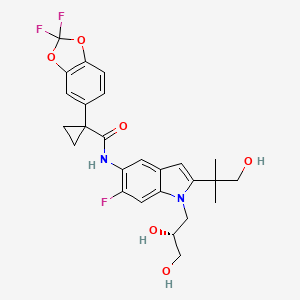



 benzhydrocodone
benzhydrocodone