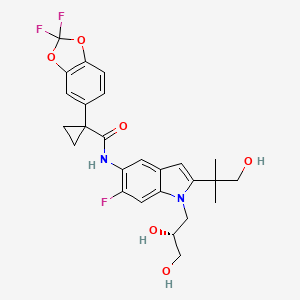
Otsuka Pharmaceutical Co., Ltd. (Otsuka) announces that the U.S. Food and Drug Administration (FDA) has approved Jynarque (tolvaptan) as the first drug treatment to slow kidney function decline in adults at risk of rapidly progressing autosomal dominant polycystic kidney disease (ADPKD).

ADPKD is a genetic disease with consequences that can lead to dialysis or kidney transplantation. It is a progressively debilitating and often painful disorder in which fluid-filled cysts develop in the kidneys over time. These cysts enlarge the kidneys and impair their ability to function normally, leading to kidney failure in most patients.3 ADPKD is diagnosed in approximately 140,000 people in the U.S.,4,5 and impacts families across multiple generations, since a parent with ADPKD has a 50 percent chance of passing the disease on to each of their children.
The efficacy of tolvaptan was demonstrated in two pivotal trials, lasting one year and three years, respectively. In the one-year REPRISE study, the primary endpoint was the treatment difference in the change of eGFR from pretreatment baseline to post-treatment follow-up, annualized by dividing by each subject’s treatment duration. In the randomized period, the change of eGFR from pretreatment baseline to post-treatment follow-up was −2.3 mL/min/1.73 m2/year with tolvaptan as compared with −3.6 mL/min/1.73 m2/year with placebo, corresponding to a treatment effect of 1.3 mL/min/1.73 m2/year (p <0.0001). In the three-year TEMPO 3:4 study, tolvaptan reduced the rate of decline in eGFR by 1.0 mL /min /1.73m2 /year (95 % confidence interval of 0.6 to 1.4) as compared to placebo in patients with earlier stages of ADPKD. In the extension trial, eGFR differences produced by the third year of the TEMPO 3:4 trial were maintained over the next 2 years of Jynarque treatment.
The primary endpoint in TEMPO 3:4 study was the intergroup difference for rate of change of total kidney volume (TKV) normalized as a percentage. The trial met its pre-specified primary endpoint of 3-year change in TKV (p<0.0001). The difference in TKV between treatment groups mostly developed within the first year, the earliest assessment, with little further difference in years two and three. In years 4 and 5 during the TEMPO 3:4 extension trial, both groups received Jynarque and the difference between the groups in TKV was not maintained. Tolvaptan has little effect on kidney size beyond what accrues during the first year of treatment. The key secondary composite endpoint (ADPKD progression) was time to multiple clinical progression events of: 1) worsening kidney function (defined as a persistent 25% reduction in reciprocal serum creatinine during treatment (from end of titration to last on-drug visit); 2) medically significant kidney pain (defined as requiring prescribed leave, last-resort analgesics, narcotic and anti-nociceptive, radiologic or surgical interventions); 3) worsening hypertension (defined as a persistent increase in blood pressure category or an increased anti-hypertensive prescription); 4) worsening albuminuria (defined as a persistent increase in albumin/creatinine ratio category). The relative rate of ADPKD-related events was decreased by 13.5% in tolvaptan-treated patients, (44 vs. 50 events per 100 person-years; hazard ratio, 0.87; 95% CI, 0.78 to 0.97; p=0.0095). As shown in the table below, the result of the key secondary composite endpoint was driven by effects on worsening kidney function and kidney pain events. In contrast, there was no effect of tolvaptan on either progression of hypertension or albuminuria. Few subjects in either arm required a radiologic or surgical intervention for kidney pain. Most kidney pain events reflected use of a medication to treat pain such as use of paracetamol, tricyclic antidepressants, narcotics and other non-narcotic agents.
Jynarque can cause serious and potentially fatal liver injury, and acute liver failure requiring liver transplantation has been reported. Jynarque has been associated with elevations of blood alanine and aspartate aminotransferases (ALT and AST), with infrequent cases of concomitant elevations in bilirubin-total (BT). To ensure the safety of patients taking Jynarque, it is necessary to measure ALT, AST and bilirubin before initiating treatment, at 2 weeks and 4 weeks after initiation, then monthly for 18 months and every 3 months thereafter, for as long as the patient is on Jynarque (tolvaptan) treatment. Because of the risks of serious liver injury, Jynarque is available only through a restricted distribution program supported by a Risk Evaluation and Mitigation Strategy (REMS) Program approved by the FDA. For more information about Jynarque, please visit www.JYNARQUE.com.
"The progressive nature of ADPKD means that kidney function gets worse over time, eventually leading to end-stage renal disease. This progression happens more rapidly for some patients than others.” said Michal Mrug, M.D., Associate Professor at the University of Alabama at Birmingham and investigator on the REPRISE trial. “Today’s approval is great news for adults at risk of rapidly progressing ADPKD because by slowing the decline in kidney function, this therapy may give them more time before kidney transplant or dialysis.”
Andy Betts, CEO of the PKD Foundation, observed, “Today is an historic day in providing hope to patients with autosomal dominant polycystic kidney disease, and we are thrilled to be a part of this first milestone in treatment. For the past 35 years, our goal has been to walk with PKD patients every step of the way. It is gratifying to play a part in the inception of the discovery of this treatment, and to see it come to fruition. We hope that this is just the beginning of a new chapter for adults at risk of rapidly progressing ADPKD who suffer from the disease.”
Also, Tatsuo Higuchi, president and representative director of Otsuka Pharmaceutical Co., Ltd., commented, “This approval is important news for many adults at risk of rapidly progressing ADPKD in the U.S., who have had no therapeutic alternatives to delay the eventual end-stage interventions of dialysis or kidney transplantation. We are humbled to be able to offer an earlier, proactive course of action to slow the progression of this disease, which we know means so much to patients, their families and healthcare providers. Simultaneously, we are grateful to the patients and researchers who through their continued commitment made this milestone possible.”




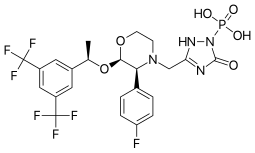

 Ivacafto
Ivacafto