Neonicotinoids-a class of neuro-active insecticides chemically similar to nicotine (family includes acetamiprid, clothianidin, imidacloprid, nitenpyram, nithiazine, thiacloprid and thiamethoxam) are currently the most widely used pesticides in the world and frequently make headlines because of their harmful effects on honeybees and other insect pollinators. Now, a study published in the prestigious journal Environmental Health Perspectives, indicates they may also have an impact on human health by disrupting our hormonal systems. This study by INRS professor Thomas Sanderson indicates that more work must be done on the potential endocrine-disrupting effects of neonicotinoids.
The Quebec government has recently decided to more strictly regulate the use of certain pesticides, including neonicotinoids, which are widely used by Quebec farmers to control crop pests. Neonicotinoids act on insects’ nervous systems, killing them by paralysis. Very little research has been done on their effects on human health, but INRS Professor Sanderson and Ph.D. student Élyse Caron-Beaudoin have taken on the challenge.
Both researchers have long been interested in the mechanisms of endocrine disrupting chemicals and they wanted to determine whether neonicotinoids belong to this class of compounds. “Endocrine disrupters are natural or synthetic molecules that can alter hormone function,” says Caron-Beaudoin, the study’s main author. ”They affect the synthesis, action, or elimination of natural hormones, which can lead to a wide variety of health effects.”
The research duo has developed methods to test the effect of neonicotinoids on the production of estrogens, essential hormones with several biological functions. By targeting aromatase, a key enzyme in the synthesis of estrogens, they were able to test the impact of three neonicotinoids on breast cancer cells in culture after exposure to concentrations similar to those found in the environment in agricultural areas.
The results of the study show an increase in aromatase expression and a unique change in the pattern in which aromatase was expressed, which is similar to that observed in the development of certain breast cancers. “However, as these results were obtained in a cellular model of breast cancer, we cannot necessarily conclude that exposure to pesticides at concentrations similar to those in the human environment would cause or promote cancer,” cautions Professor Sanderson. This study is the first evidence that neonicotinoids have an effect on aromatase gene expression and may potentially alter estrogen production. Hormonal disturbance by these pesticides will need to be confirmed in future studies, but the results obtained by the INRS team indicate that it caution should be exercised in the management and use of neonicotinoid insecticides.
Ref : https://ehp.niehs.nih.gov/EHP2698/





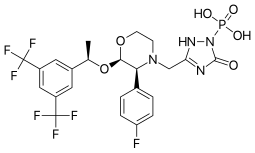

 Ivacafto
Ivacafto