The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive).
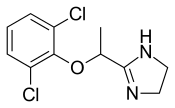
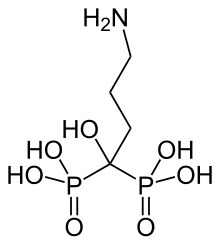
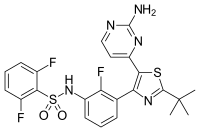 dabrafenib
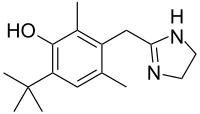
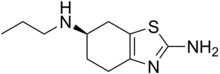
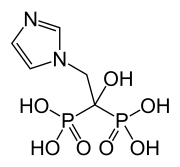
dabrafenib  trametinib
trametinib
“This is the first FDA-approved treatment for patients with this aggressive form of thyroid cancer, and the third cancer with this specific gene mutation that this drug combination has been approved to treat,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients.”
Thyroid cancer is a disease in which cancer cells form in the tissues of the thyroid gland. Anaplastic thyroid cancer is a rare, aggressive type of thyroid cancer. The National Institutes of Health estimates there will be 53,990 new cases of thyroid cancer and an estimated 2,060 deaths from the disease in the United States in 2018. Anaplastic thyroid cancer accounts for about 1 to 2 percent of all thyroid cancers.
Both Tafinlar and Mekinist are also approved for use, alone or in combination, to treat BRAF V600 mutation-positive metastatic melanoma. Additionally, Tafinlar and Mekinist are approved for use, in combination, to treat BRAF V600E mutation-positive, metastatic non-small cell lung cancer.
The efficacy of Tafinlar and Mekinist in treating ATC was shown in an open-label clinical trial of patients with rare cancers with the BRAF V600E mutation. Data from trials in BRAF V600E mutation-positive, metastatic melanoma or lung cancer and results in other BRAF V600E mutation-positive rare cancers provided confidence in the results seen in patients with ATC. The trial measured the percent of patients with a complete or partial reduction in tumor size (overall response rate). Of 23 evaluable patients, 57 percent experienced a partial response and 4 percent experienced a complete response; in nine (64 percent) of the 14 patients with responses, there were no significant tumor growths for six months or longer.
The side effects of Tafinlar and Mekinist in patients with ATC are consistent with those seen in other cancers when the two drugs are used together. Common side effects include fever (pyrexia), rash, chills, headache, joint pain (arthralgia), cough, fatigue, nausea, vomiting, diarrhea, myalgia (muscle pain), dry skin, decreased appetite, edema, hemorrhage, high blood pressure (hypertension) and difficulty breathing (dyspnea).
Severe side effects of Tafinlar include the development of new cancers, growth of tumors in patients with BRAF wild-type tumors, serious bleeding problems, heart problems, severe eye problems, fever that may be severe, serious skin reactions, high blood sugar or worsening diabetes, and serious anemia.
Severe side effects of Mekinist include the development of new cancers; serious bleeding problems; inflammation of intestines and perforation of the intestines; blood clots in the arms, legs or lungs; heart problems; severe eye problems; lung or breathing problems; fever that may be severe; serious skin reactions; and high blood sugar or worsening diabetes.
Both Tafinlar and Mekinist can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted Priority Review and Breakthrough Therapy designation for this indication. Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases, was also granted for this indication.