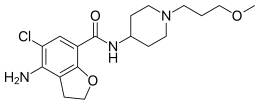
SHPG) has announced that the U.S. Food and Drug Administration (FDA) has approved Motegrity (prucalopride), a once-daily, oral treatment option for adults with Chronic Idiopathic Constipation (CIC).
Motegrity, a selective serotonin-4 (5-HT4) receptor agonist, provides a different class of treatment for CIC that works by enhancing colonic peristalsis to increase bowel motility. Motegrity is expected to launch in 2019 in the United States, where an estimated 35 million adults are living with CIC. While not all patients may be right for treatment, Motegrity represents an important new option.
“The approval of Motegrity marks a new day in the treatment of CIC,” said Howard Mayer, M.D., Senior Vice President and Chief Medical Officer, Shire. “This significant milestone reinforces our continued commitment to the GI community and advances our goal of addressing the unmet need of patients suffering from rare, specialized and common GI conditions.”
The efficacy of once-daily treatment with Motegrity was evaluated in six double-blind, placebo-controlled, randomized, multicenter clinical studies lasting 12 weeks (studies 1-5) or 24 weeks (study 6) Of the 2,484 patients, most were female (76%) and Caucasian (76%), with an average age of 47 (+/- 16 years).
“As a gastroenterologist, it’s important for me to help patients with CIC find a treatment that works well for them,” said Brooks Cash, M.D., Chief of the Division of Gastroenterology, Hepatology, and Nutrition at the University of Texas Health Science Center at Houston. “It’s exciting to be able to now offer my patients a new treatment option that addresses colonic peristalsis.”
During studies, significantly more patients taking Motegrity achieved the primary endpoint (an average of ≥3 complete spontaneous bowel movements [CSBMs] per week over 12 weeks, considered normalization of BM frequency) than those in the placebo group (19-38% Motegrity ≤2 mg vs. 10-20% placebo) across five of six trials. A rapid response was seen with Motegrity as early as week 1, with improvements maintained throughout 12 weeks of treatment.1 The FDA has requested that Shire conduct five post-marketing studies evaluating the pharmacokinetics, efficacy, and safety of Motegrity in pediatric patients with CIC (6 months old to less than 18 years of age) and pregnant and lactating women with CIC treated with Motegrity.
Motegrity is contraindicated in patients with a history of hypersensitivity to Motegrity. Reactions include dyspnea, rash, pruritus, urticaria, and facial edema have been observed. Motegrity is also contraindicated in patients with intestinal perforation or obstruction due to structural or functional disorder of the gut wall, obstructive ileus, severe inflammatory conditions of the intestinal tract such as Crohn’s disease, ulcerative colitis, and toxic megacolon/megarectum.
In clinical trials, suicides, suicide attempts, and suicidal ideation have been reported. A causal association between treatment with Motegrity and an increased risk of suicidal ideation and behavior has not been established. Monitor all patients treated with Motegrity for persistent worsening of depression or the emergence of suicidal thoughts and behaviors. Counsel patients, their caregivers, and family members of patients to be aware of any unusual changes in mood or behavior and alert the healthcare provider. Instruct patients to discontinue Motegrity immediately and contact their healthcare provider if they experience any of these symptoms.
Most common adverse reactions (≥2%) are headache, abdominal pain, nausea, diarrhea, abdominal distension, dizziness, vomiting, flatulence and fatigue. Overall, discontinuation due to adverse events was low (5% Motegrity 2 mg once daily; 3% placebo). If reported, adverse events of diarrhea or headache typically resolved within a few days.1 In addition, cardiovascular safety was evaluated in a MACE† (major adverse cardiovascular events) analysis of the double-blind, placebo-controlled and open-label studies. It was also assessed in a retrospective observational study, which demonstrated no increase in the risk of MACE with Motegrity relative to polyethylene glycol (PEG).
CIC is a common condition affecting roughly 14% of the adult population. Symptoms can range from straining and bloating, to infrequent, or incomplete bowel movements. While “idiopathic” by definition (meaning the exact cause is not known), it is believed that CIC may be caused by insufficient movement of the colon muscle
------------------------------------------------------------------------------------------------------------------------