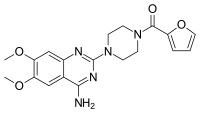
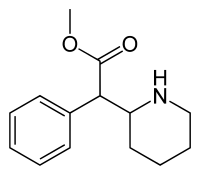
In continuation of my update on Remoxy ER (oxycodone)
Pain Therapeutics, Inc. (Nasdaq: PTIE), a clinical-stage drug development company, today announced feedback from a meeting held January 31, 2019 with the U.S. Food and Drug Administration (FDA) regarding the drug candidate, Remoxy ER. Remoxy is the trade name for a new type of abuse-deterrent, extended-release gel formulation of oxycodone (CII) with physical/chemical properties intended to deter abuse. As previously disclosed, we requested this meeting to resolve disagreement around comments and conclusions made by FDA in 2018 during a regulatory review of a New Drug Application (NDA) for Remoxy.
During this meeting, we learned that i) FDA denies making math errors, material mistakes or misrepresentations during a June 2018 Advisory Committee Meeting for Remoxy, despite clear evidence to the contrary; ii) comparator data is irrelevant for the evaluation of abuse-deterrent properties, despite FDA written guidance which explicitly states the opposite; and (ii) that we would need to rely on the Freedom of Information Act to access additional data generated by FDA with Remoxy. As a result of our recent meeting with FDA, we believe we are no closer today to product approval than we were over a year ago.
“Remoxy remains an odyssey without a homecoming,” said Remi Barbier, President & CEO of Pain Therapeutics. “We had hoped for a fair, neutral and impartial review of the Remoxy data. Instead, we walked out of this meeting feeling a bit disoriented by FDA’s lack of transparency, clarity or helpfulness. It’s a rare occasion when two parties can’t agree on simple math. We can’t work with shambolic regulations. This is not how you win support for innovation.”
Historically, the lead candidate in our pipeline has been Remoxy, an analgesic drug that we conceived, patented, developed and tested in collaboration with corporate and academic partners. Over the years, we have conducted a successful clinical development program for Remoxy, including a large, well-controlled pivotal Phase III efficacy study whose primary endpoints met statistical significance (p<0.05). The clinical safety or analgesic efficacy of Remoxy for its intended purpose is not in question. Its abuse-deterrent properties, however, are subject of a difference of opinion. Abuse deterrence refers to properties that are embedded into an opioid formulation to prevent certain common methods of abuse. During the long development history of Remoxy, we generated nearly 9,000 unique data points in over 50 studies at a cost in excess of $100 million. Studies were designed in consultation with FDA and conducted by independent labs. Collectively, we believe these studies adequately characterize Remoxy’s abuse-deterrent properties. In particular, we demonstrated that the two currently marketed extended-release oxycodone products -- OxyContin® and Xtampza® -- which both benefit from abuse-deterrent label claims, can both be defeated for purposes of abuse in under a minute using common household items. In contrast, Remoxy requires a significant investment of time, effort and equipment to defeat, and even then, results in less release of oxycodone. During our recent meeting with FDA we were informed they believe Remoxy capsules lack abuse deterrence via the injection route of abuse because “oxycodone can be extracted from the product”, regardless of how much time, effort, frustration or equipment is required to so do. We are unable to follow the logic by which a drug product should never release drug. More generally, as the regulatory requirements for Remoxy have changed frequently and suddenly over time, we have experienced significant delays and have incurred unanticipated expenses related to the overall Remoxy development program.
-more-
We believe innovative products such as Remoxy can serve a meaningful social purpose and, potentially, may save lives during the worst drug crisis in American history. By necessity, however, we rely on reasonably predictive regulatory pathways to guide our product candidates through development in preparation for commercialization. We also rely on principles of good governance, in which similar drugs receive similar regulatory treatment under rules that are clear, publicized, and evenly applied. In our experience with Remoxy, the regulatory environment around abuse-deterrence lacks these essential qualities.
There are procedures in place at the FDA and other government agencies to help promote a fair resolution of disputes. Such procedures can be complex and may not be rapid, predictable or even viable. Going forward, we will generally be silent regarding our plans or future expectations for Remoxy, unless a significant material event occurs that compels us to update our public disclosures around this product candidate.
https://www.drugbank.ca/drugs/DB00497
https://medlineplus.gov/druginfo/meds/a682132.html