In continuation f my update on ALKS 5461
Alkermes plc (Nasdaq: ALKS) announced that it received a Complete Response Letter (CRL) from the U.S. Food and Drug Administration (FDA) regarding its New Drug Application (NDA) for ALKS 5461 for the adjunctive treatment of major depressive disorder (MDD).
The CRL states that the FDA is unable to approve the ALKS 5461 NDA in its present form and is requesting additional clinical data to provide substantial evidence of effectiveness of ALKS 5461 for the adjunctive treatment of MDD. Alkermes plans to meet with the FDA to discuss the contents of the CRL and potential next steps for ALKS 5461. This interaction with the Agency will inform whether there is a viable path forward for the ALKS 5461 program.
The NDA submission for ALKS 5461 was based on results from a clinical efficacy and safety package with data from more than 30 clinical trials and more than 1,500 patients with MDD. Throughout the clinical development program, ALKS 5461 demonstrated a consistent profile of antidepressant activity, safety and tolerability in the adjunctive treatment of MDD.
About ALKS 5461
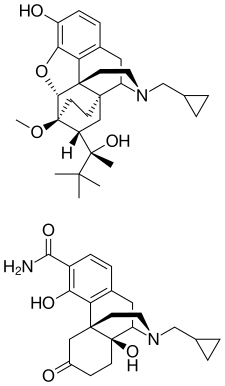
ALKS 5461 is a proprietary, investigational, once-daily oral medicine that acts as an opioid system modulator and represents a novel mechanism of action for the adjunctive treatment of major depressive disorder (MDD) in patients with an inadequate response to standard antidepressant therapies. ALKS 5461 is a fixed-dose combination of buprenorphine, a partial mu-opioid receptor agonist and kappa-opioid receptor antagonist, and samidorphan, a mu-opioid receptor antagonist.
About Major Depressive Disorder (MDD)
According to the DSM-5® (Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition), major depressive disorder (MDD) is a condition in which patients exhibit depressive symptoms, such as a depressed mood or a loss of interest or pleasure in daily activities consistently for at least a two-week period, and demonstrate impaired social, occupational, educational or other important functioning. An estimated 16.2 million people in the U.S. suffered from MDD in 2016,1 the majority of whom may not adequately respond to initial antidepressant therapy.
https://en.wikipedia.org/wiki/Buprenorphine/samidorphan