Bayer announced the completion of the rolling submission of a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for the investigational drug darolutamide. The submission, which was initiated in December 2018, is based on data from the Phase III ARAMIS trial in men with non-metastatic castration-resistant prostate cancer (nmCRPC).1 These data were recently presented at the American Society of Clinical Oncology Genitourinary Cancers Symposium (ASCO GU) in San Francisco and published simultaneously in The New England Journal of Medicine.
"We are grateful to the patients, their families and the clinical investigators who have made this important study possible," said Scott Z. Fields, M.D., senior vice president and head of Oncology Development at Bayer's Pharmaceutical Division. "The NDA submission is a key milestone bringing us closer to providing darolutamide as a potential treatment option for men with nmCRPC."
Bayer has been granted Fast Track designation by the FDA for darolutamide in men with nmCRPC. Bayer is also in discussions with other health authorities regarding a submission of darolutamide. The compound is being developed jointly by Bayer and Orion Corporation, a globally operating Finnish pharmaceutical company.
About the ARAMIS Trial
The ARAMIS trial is a randomized, Phase III, multi-center, double-blind, placebo-controlled trial evaluating the safety and efficacy of oral darolutamide in patients with nmCRPC who are currently being treated with androgen deprivation therapy (ADT) as standard of care and are at high risk for developing metastatic disease. 1,509 patients were randomized in a 2:1 ratio to receive 600 mg of darolutamide twice a day or placebo along with ADT.
The primary endpoint of this trial is metastasis-free survival (MFS) defined as time between randomization and evidence of metastasis or death. The secondary endpoints of this trial are overall survival (OS), time to pain progression, time to initiation of first cytotoxic chemotherapy, time to first symptomatic skeletal event (SSE), and characterization of the safety and tolerability of darolutamide.
About Darolutamide
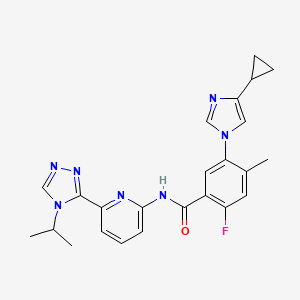
Darolutamide is an investigational, non-steroidal androgen receptor antagonist with a chemical structure that binds to the receptor and exhibits antagonistic activity, thereby inhibiting the receptor function and the growth of prostate cancer cells. A Phase 3 study in metastatic hormone-sensitive prostate cancer (ARASENS) is ongoing. Information about these trials can be found at www.clinicaltrials.gov.
Darolutamide is not approved by the U.S. FDA, the European Medicines Agency or any other health authority.
https://en.wikipedia.org/wiki/Darolutamide