Alkermes plc (Nasdaq: ALKS) and Biogen Inc. (Nasdaq: BIIB) announced that the U.S. Food and Drug Administration (FDA) has accepted for review the New Drug Application (NDA) for diroximel fumarate (BIIB098), a novel oral fumarate in development for the treatment of relapsing forms of multiple sclerosis (MS). The NDA has been assigned a PDUFA (Prescription Drug User Fee Act) target action date in the fourth quarter of 2019. If approved, Biogen intends to market diroximel fumarate under the brand name VUMERITY™, which has been conditionally accepted by the FDA and will be confirmed upon approval.
“The NDA filing acceptance for diroximel fumarate further demonstrates the productive collaboration between Alkermes and Biogen and brings us closer to our shared goal of offering a new therapeutic option for people with MS,” said Craig Hopkinson, M.D., chief medical officer and senior vice president, medicines development and medical affairs at Alkermes. “We believe diroximel fumarate has the potential to be a meaningful new offering for patients with MS, and we look forward to continued engagement with the FDA throughout the review process.”
“For more than two decades Biogen has been at the forefront of delivering new medicines to MS patients,” said Michael Ehlers, M.D., Ph.D., executive vice president, research and development at Biogen. “We are encouraged by the FDA’s acceptance of the NDA for diroximel fumarate, which we believe could help elevate the treatment of this complex and often debilitating disease.”
Alkermes is seeking approval of diroximel fumarate under the 505(b)(2) regulatory pathway, referencing Biogen’s dimethyl fumarate data. The NDA submission includes data from EVOLVE-MS-1, a Phase 3, open-label, two-year safety study in relapsing-remitting MS (RRMS) patients. It is hypothesized that the distinct chemical structure of diroximel fumarate may impact its gastrointestinal (GI) tolerability. Alkermes is conducting EVOLVE-MS-2, a head-to-head GI tolerability study versus dimethyl fumarate, with results expected later this year.
About the Diroximel Fumarate Clinical Development Program
The key components of the clinical development program of diroximel fumarate include the EVOLVE-MS-1 study, a Phase 3, open-label, two-year safety study in relapsing-remitting MS (RRMS) patients, along with pharmacokinetic bridging studies comparing diroximel fumarate and dimethyl fumarate. In addition, Alkermes is conducting the EVOLVE-MS-2 study in patients with RRMS, a five-week, head-to-head gastrointestinal (GI) tolerability study versus dimethyl fumarate.
About Diroximel Fumarate
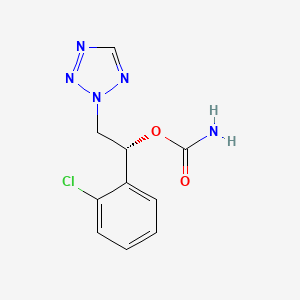
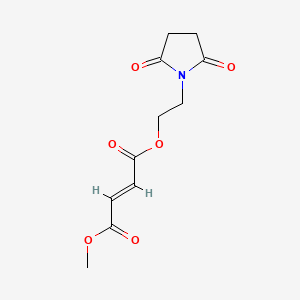
Diroximel fumarate (BIIB098) is a novel oral fumarate candidate in development for the treatment of relapsing forms of MS. Diroximel fumarate is designed to rapidly convert to monomethyl fumarate in the body and may have the potential to offer differentiated GI tolerability due to its chemical structure as compared to dimethyl fumarate.