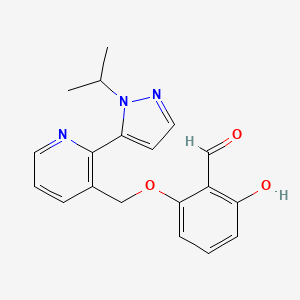
In continuation of my update on Selinexor
Karyopharm Therapeutics Inc. an oncology-focused pharmaceutical company, announced that the U.S. Food and Drug Administration (FDA) has approved oral Xpovio (selinexor), a nuclear export inhibitor, in combination with dexamethasone for the treatment of adult patients with relapsed or refractory multiple myeloma (RRMM) who have received at least four prior therapies and whose disease is refractory to at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody. This indication is approved under accelerated approval based on response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. The ongoing, randomized Phase 3 BOSTON study evaluating selinexor in combination with Velcade® (bortezomib) and low-dose dexamethasone will serve as the confirmatory trial. The FDA’s Accelerated Approval Program was developed to allow for expedited approval of drugs that treat serious conditions and that fill an unmet medical need.
Karyopharm expects Xpovio to become commercially available in the U.S. on or before July 10, 2019. A Marketing Authorization Application for selinexor is also currently under review by the European Medicines Agency.
“With today’s accelerated approval of Xpovio by the FDA, patients with heavily pretreated multiple myeloma will now have a new therapeutic option to treat their disease,” said Sharon Shacham, PhD, MBA, Founder, President and Chief Scientific Officer of Karyopharm. “Discovering, developing and securing FDA approval for XPOVIO with its novel mechanism of action over the past decade required the dedication of many people, including the patients, caregivers and physicians involved in our clinical trials, along with the many employees at Karyopharm. We are tremendously grateful for everyone’s contributions to this important milestone, and we look forward to the next stage in our pursuit of improving the lives of patients with cancer.”
“The 25.3% response rate seen in the subgroup of 83 patients in the pivotal Phase 2b STORM study that served as the basis for Xpovio's accelerated approval is clinically meaningful and a validated surrogate marker for clinical benefit in our patients with advanced refractory disease,” said Sundar Jagannath, MD, Director of the Multiple Myeloma Program, Professor of Medicine (Hematology and Medical Oncology) at Tisch Cancer Institute at Mount Sinai School of Medicine, and principal investigator of the STORM study.
“Despite recent advances in the treatment of multiple myeloma, almost all our patients will develop disease that is resistant to the five most commonly used anti-myeloma drugs we currently have available, and the prognosis for this patient population is particularly poor. The accelerated approval of oral Xpovio marks an important advance in the treatment paradigm for patients with relapsed refractory multiple myeloma, and in my view, is an important addition to our therapeutic armamentarium,” said Dr. Paul Richardson, MD, Clinical Program Leader and Director of Clinical Research, Jerome Lipper Multiple Myeloma Center at the Dana-Farber Cancer Institute.
Michael G. Kauffman, MD, PhD, Chief Executive Officer of Karyopharm, commented, “Having worked on novel drugs in myeloma beginning with Velcade in the year 2000, I have been thrilled to see such exciting progress overall in the field where there are substantial increases in patients’ duration and quality of life. The accelerated approval of oral Xpovio targeting XPO1 represents the first approval against a new target in myeloma since 2015, and we look forward to advancing the further clinical development of Xpovio.”
https://pubchem.ncbi.nlm.nih.gov/compound/Selinexor