Kyowa Kirin Co., Ltd., (Kyowa Kirin, TYO: 4151) announced the U.S. Food and Drug Administration (FDA) has granted approval for Nourianz (istradefylline) for use as adjunctive treatment to levodopa/carbidopa in adult patients with Parkinson’s disease (PD) experiencing “OFF” episodes.
“We are proud that Nourianz is now ready to help adult patients with Parkinson’s disease in the US,” said Tomohiro Sudo, Head of Global Product Management Office of Kyowa Kirin. “We believe that Nourianz could be an important contributor to improve treatment outcomes. We will keep working to bring the product to patients globally.”
“Kyowa Kirin has a commitment to global health and well-being by creating new value through the pursuit of advances in life sciences and technology particularly in oncology, nephrology, immunology, and the central nervous system,” said Tom Stratford, President of Kyowa Kirin USA Holdings, Inc. “Today's FDA approval of Nourianz is an important milestone and provides US patients with a novel non-dopaminergic once-a-day oral treatment option to be used in conjunction with levodopa/carbidopa for Parkinson’s disease.”
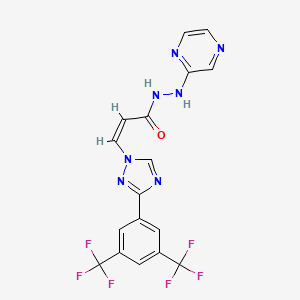
“Today’s approval is the culmination of decades of perseverance in exploring the science and clinical effects of istradefylline and inhibition of adenosine A2A receptor signaling in people with Parkinson’s disease,” said Jeffrey S. Humphrey, MD, Chief Development Officer of Kyowa Kirin Pharmaceutical Development, Inc. “In clinical studies, istradefylline, used as adjunctive treatment to levodopa/carbidopa in adult patients with PD experiencing 'OFF' episodes, was associated with a decrease in OFF Time and increase in ON Time without troublesome dyskinesia. We are grateful for the FDA approval and for the many dedicated scientists and patients whose participation in our research programs has resulted in a new treatment option for Parkinson's disease.”
“Istradefylline is an Adenosine A2A receptor antagonist, and is a novel non-dopaminergic pharmacologic approach to treating OFF episodes for people living with PD,” said Dr. Stuart Isaacson, MD, Parkinson’s Disease and Movement Disorders Center of Boca Raton, Florida. “Based on data from four clinical studies, istradefylline taken as an adjunct to levodopa significantly improved OFF time and demonstrated a well-tolerated safety profile. Istradefylline represents an important new treatment option for patients with Parkinson's disease who experience 'OFF' episodes.”
The FDA approval of Nourianz is based on findings from randomized, multi-center, double-blind, placebo-controlled trials in patients with PD taking a stable dose of levodopa/carbidopa with or without other PD medications.
The Kyowa Kirin Group companies strive to contribute to the health and well-being of people around the world by creating new value through the pursuit of advances in life sciences and technologies.
Please see Nourianz indication and Important Safety Information below.
Indication
Nourianz (istradefylline) is an adenosine receptor antagonist indicated as adjunctive treatment to levodopa/carbidopa in adult patients with Parkinson’s disease (PD) experiencing “off” episodes.
Important Safety Information
Warnings and Precautions
Dyskinesia: Nourianz in combination with levodopa may cause dyskinesia or exacerbate pre-existing dyskinesia. In clinical trials, 1% of patients treated with either Nourianz 20 mg or 40 mg discontinued treatment because of dyskinesia, compared to 0% for placebo.