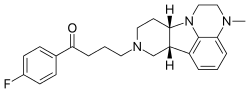
BeiGene, Ltd. (NASDAQ: BGNE; HKEX: 06160), a commercial-stage biopharmaceutical company focused on developing and commercializing innovative molecularly-targeted and immuno-oncology drugs for the treatment of cancer, announced that Brukinsa (zanubrutinib) has received accelerated approval from the United States Food and Drug Administration (FDA) as a treatment for mantle cell lymphoma (MCL) in adult patients who have received at least one prior therapy. Brukinsa is the first BeiGene-discovered product to be approved, an important milestone toward the company’s goal of transforming treatment for cancer patients around the world.
This accelerated approval is based on overall response rate (ORR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
“We are working to improve outcomes for people with cancer worldwide and this approval brings us closer to realizing our mission of bringing the highest quality therapies to patients globally,” said John V. Oyler, Chairman, Co-Founder, and CEO of BeiGene. “Today’s FDA approval of Brukinsa, following the previously granted Breakthrough Therapy designation in this indication, validates it as an important treatment option for people with relapsed or refractory MCL. We hope this is the first of many approvals for Brukinsa as we continue to evaluate its potential in other hematologic cancers.”
“Brukinsa is a BTK inhibitor that was designed to maximize target occupancy and minimize off-target binding. It entered the clinic in 2014 and since that time our broad development program has enrolled more than 1,600 patients globally,” said Jane Huang, M.D., Chief Medical Officer, Hematology at BeiGene. “Today’s accelerated approval is the culmination of many years of effort by the BeiGene team, the dedicated investigators involved in these trials and, most importantly, the patients who participated by enrolling in the clinical trials. We are humbled by the opportunity to develop this therapy and launch it as our first internally discovered and approved cancer treatment.”
“BTK inhibition is an established mode of treatment for patients with MCL, but many patients treated with previously approved BTK inhibitors do not fully respond to BTK therapy or are forced to discontinue treatment early due to side effects. Today we have a new option for our adult patients who have received one prior systemic or targeted therapy and are living with MCL, an aggressive blood cancer that’s often diagnosed at a more advanced stage,” said Luhua (Michael) Wang, M.D., Professor, Department of Lymphoma and Myeloma, Division of Cancer Medicine at The University of Texas MD Anderson Cancer Center, and clinical trial investigator.
“The approval of Brukinsa as a second line therapy represents an important advancement for the treatment of mantle cell lymphoma,” said Meghan Gutierrez, Chief Executive Officer for the Lymphoma Research Foundation. “Expanded treatment options can transform the patient experience and provide hope to people living with a mantle cell diagnosis.”
The FDA’s approval of Brukinsa is based on efficacy results from two single-arm clinical trials, with independent review committee (IRC)-assessed ORR per 2014 Lugano Classification as the primary endpoint. Across both trials, Brukinsa achieved an ORR, which is the sum of complete responses and partial responses, of 84%.
In the multicenter Phase 2 trial of zanubrutinib in patients with relapsed or refractory (R/R) MCL BGB-3111-206 (NCT03206970), the ORR was 84% (95% CI: 74%, 91%), including 59% complete response (FDG-PET scan required) and 24% partial response. In this study, the median duration of response (DOR) was 19.5 months (95%CI: 16.6, NE) and median follow-up time on study was 18.4 months. In the global Phase 1/2 trial BGB-3111-AU-003 (NCT02343120), the ORR was 84% (95% CI: 67%, 95%), including 22% complete response (FDG-PET scan not required) and 62% partial response. In this study, the median DOR was 18.5 months1 (95% CI:12.6, NE) and median follow-up time on study was 18.8 months.
The most common adverse reactions (> 10%) with Brukinsa were decreased neutrophil count, decreased platelet count, upper respiratory tract infection, decreased white blood cell count, decreased hemoglobin, rash, bruising, diarrhea, cough, musculoskeletal pain, pneumonia, urinary tract infection, blood in the urine (hematuria), fatigue, constipation, and hemorrhage. The most frequent serious adverse reactions were pneumonia (11%) and hemorrhage (5%).
Of the 118 patients with MCL treated with Brukinsa, eight (7%) patients discontinued treatment due to adverse reactions in the trials. The most frequent adverse reaction leading to treatment discontinuation was pneumonia (3.4%). One (0.8%) patient experienced an adverse reaction leading to dose reduction (hepatitis B).
The recommended dose of Brukinsa is 320 mg, taken orally 160 mg twice daily or 320 mg once daily with or without food. The dose may be adjusted for adverse reactions, and reduced for patients with severe hepatic impairment and certain drug interactions.
https://en.wikipedia.org/wiki/Zanubrutinib