Researchers from Case Western Reserve University School of Medicine, University Hospitals Cleveland Medical Center (UH), Cleveland Clinic and Lifebanc (a Northeast Ohio organ-procurement organization) have developed a new way to preserve donated kidneys—a method that could extend the number and quality of kidneys available for transplant, saving more people with end-stage renal disease, more commonly known as "kidney failure."
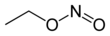

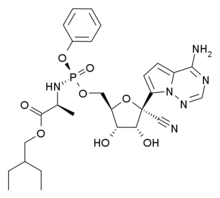
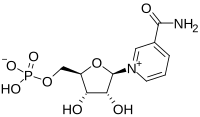
The team identified a drug—ethyl nitrite-See structure 1—that could be added to the preservation fluid to generate tiny molecules called S-nitrosothiols (SNOs) see structure 2, which regulate tissue-oxygen delivery. This, in turn, restored flow-through and reduced resistance within the kidney. Higher flow-rates and lower resistance are associated with better kidney function after transplantation.
Their research was funded by a grant from the Roche Organ Transplant Research Foundation and recently published in Annals of Surgery.
The United States has one of the world's highest incidences of end-stage renal disease, and the number of afflicted individuals continues to increase. The prevalence of end-stage renal disease has more than doubled between 1990 and 2016, according to the Centers for Disease Control.
The optimal treatment is a kidney transplant, but demand far exceeds supply. Additionally, donation rates for deceased donors have been static for several years, despite various public-education campaigns, resulting in fewer kidneys available for transplant. And while the proportion and number of living donors has increased, this latter group still only makes up a small percentage of recovered kidneys for transplant.
Increasing the number of kidneys available for transplant benefits patients by extending lifespans and/or enhancing quality of life as well as the potential for reducing medical costs (a transplant is cheaper than ongoing dialysis). To help improve outcomes for kidney transplant patients, the team explored ways to extend the viability of donated kidneys.
Improvements in surgical techniques and immunosuppression therapies have made kidney transplants a relatively common procedure. However, less attention has been paid to maintaining/improving kidney function during the kidney-transport phase.
"We addressed this latter point through developing enhanced preservation methods," said senior author James Reynolds, professor of Anesthesiology and Perioperative Medicine at Case Western Reserve School of Medicine and a member of the Harrington Discovery Institute at UH.
For decades, procured kidneys were simply flushed with preservation solution and then transported in ice-filled coolers to the recipient's hospital. But advances in pumping technology slowly changed the field toward active storage, the preferred method for conveying the organ from donor to recipient.
"However, while 85% of kidneys are now pumped, up to 20% of kidneys are determined to be unsuitable for transplant during the storage phase," said Kenneth Chavin, professor of surgery at the School of Medicine, chief of hepatobiliary and transplant surgery and director of the UH Transplant Institute.
"For several years, our team has directed research efforts toward understanding and improving the body's response to medical manipulation," Reynolds said. "Organ-donor physiology and 'transport status' fit well within this metric. We identified a therapy that might improve kidney perfusion, a significant factor in predicting how the organ will perform post-transplant."
Previous work by Reynolds and long-time collaborator Jonathan Stamler, the Robert S. and Sylvia K. Reitman Family Foundation Distinguished Chair in Cardiovascular Innovation and president of the Harrington Discovery Institute, determined that brain death significantly reduces SNOs, which impairs blood-flow and tissue-oxygenation to the kidneys and other commonly transplanted organs. The loss of SNOs is not corrected by current preservation fluids, so impaired flow through the kidneys continues during storage and transport.
https://journals.lww.com/annalsofsurgery/Abstract/publishahead/A_Novel_Method_to_Improve_Perfusion_of_Ex_Vivo.94769.aspx
https://en.wikipedia.org/wiki/Ethyl_nitrite
https://en.wikipedia.org/wiki/S-Nitrosothiol