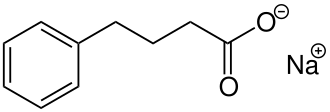
Acer Therapeutics Inc. and its collaboration partner, RELIEF THERAPEUTICS Holding SA (“Relief”), announced the U.S. Food and Drug Administration (FDA) approval Olpruva™ (sodium phenylbutyrate) of oral suspension in the U.S. for the treatment of certain patients living with urea cycle disorders (UCDs) involving deficiencies of carbamylphosphate synthetase (CPS), ornithine transcarbamylase (OTC), or argininosuccinic acid synthetase (AS).
“The FDA’s approval of Olpruva, an innovative formulation of sodium phenylbutyrate packaged for the first time in single-dose envelopes, marks the culmination of our ongoing dedication to develop new and differentiated treatment options for those affected by rare diseases,” said Chris Schelling, chief executive officer and founder of Acer. “Patients who are living with UCDs now have an alternative treatment option with Olpruva™, to address some of the challenges they may have with existing therapy. We are pleased to be able to provide a new, approved treatment choice for those living with this challenging disease.”
Mr. Schelling continued, “This approval represents the first FDA-approved product for Acer, validating our ability to identify and develop treatments where science can be applied in novel ways and make them available to patients as quickly and efficiently as possible. In addition, this approval unlocks our Marathon debt funding option and provides us with resources to advance our pipeline of investigational product candidates.”
“The daily challenges of living with a UCD can be overwhelming and emotionally draining for patients and their families,” said Tresa Warner, president of the National Urea Cycle Disorders Foundation. “We welcome new treatment options that can help patients, caregivers and their healthcare teams manage UCDs.”
Olpruva’s™ approval triggers the availability of a $42.5 million term loan to Acer under the previously announced March 2022 loan agreement the Company entered into with affiliates of Marathon Asset Management L.P. If Acer requests and receives the loan proceeds, management believes it will have sufficient resources to fund current operations into H2 2023.
Olpruva™ has been approved as an oral suspension by the FDA for the treatment of patients with UCDs. UCDs are a group of rare, genetic disorders that can cause harmful ammonia to build up in the blood, potentially resulting in brain damage and neurocognitive impairments, if ammonia levels are not controlled.1 Any increase in ammonia over time is serious. Therefore, it is important to adhere to any dietary protein restrictions and have alternative medication options to help control ammonia levels.
“This FDA approval is a significant milestone for patients with UCDs in the U.S., offering an additional choice to manage their condition,” added Jack Weinstein, chief executive officer of Relief. “We look forward to building on Olpruva™’s approval in the U.S. and expanding its availability into other territories outside of the U.S.”
Olpruva™ received FDA approval under section 505(b)(2) of the Federal Food, Drug and Cosmetic Act (FDCA), a regulatory pathway that allows applicants to rely, at least in part, on third party data for approval. In its New Drug Application (NDA), Acer cited preclinical and clinical safety and efficacy data from the reference listed drug (RLD), BUPHENYL® powder, which is approved as adjunctive therapy in the chronic management of patients with UCDs involving deficiencies of CPS, OTC or AS. In its NDA, Acer also provided additional data including studies that evaluated the bioavailability and bioequivalence of Olpruva™ compared to BUPHENYL® powder. The data from these studies, presented at the Society for Inherited Metabolic Disorders (SIMD) Annual Meeting in April 2022 and the Genetic Metabolic Dieticians International (GMDI) Conference in May 2022, showed that Olpruva™ was bioequivalent to BUPHENYL® powder.