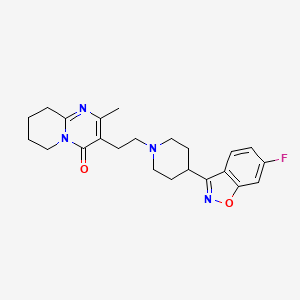
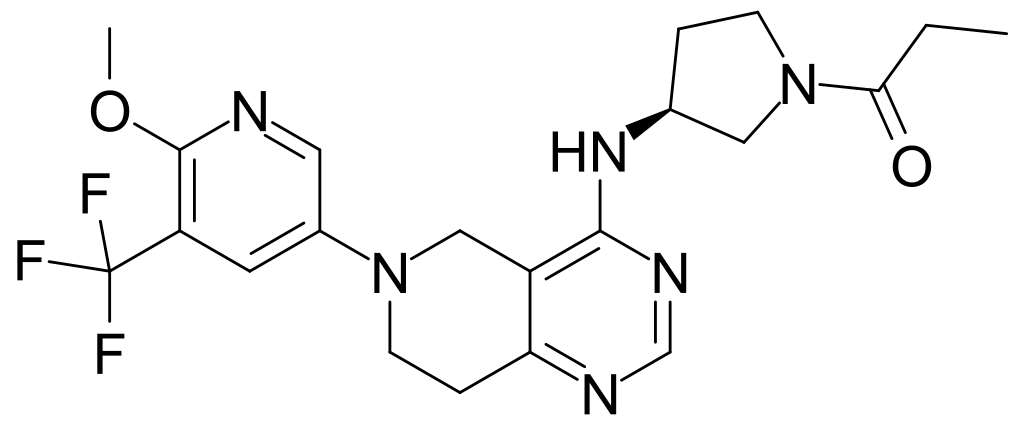
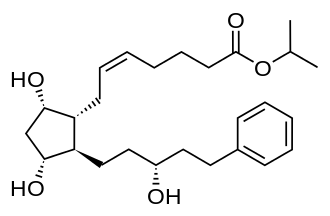
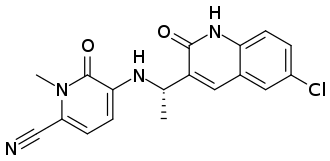
Neurofilaments are proteins that are released from neurons when they are damaged, making them a marker of neurodegeneration.6
“For more than a decade, Biogen has been steadfast in our commitment to pursuing treatments for ALS, and I want to thank the scientists as well as the entire ALS community who have all worked tirelessly to bring this first-of-its-kind treatment to people with SOD1-ALS,” said Christopher A. Viehbacher, President and Chief Executive Officer of Biogen. “Today also marks a pivotal moment in ALS research as we gained, for the first time, consensus that neurofilament can be used as a surrogate marker reasonably likely to predict clinical benefit in SOD1-ALS. We believe this important scientific advancement will further accelerate innovative drug development for ALS.”
Qalsody is the first approved treatment to target a genetic cause of ALS. Biogen collaborated with Ionis Pharmaceuticals on the early development of tofersen.
Warnings and precautions associated with Qalsody were serious neurologic events, including myelitis and/or radiculitis; papilledema and elevated intracranial pressure; and aseptic meningitis. If symptoms consistent with myelitis, radiculitis papilledema, elevated intracranial, or aseptic meningitis develop, diagnostic workup and treatment should be initiated according to the standard of care. Management may require interruption or discontinuation of Qalsody. The most common adverse reactions that occurred in ≥10% of Qalsody treated participants and more than the placebo arm were pain, fatigue, arthralgia, cerebrospinal (CSF) white blood cell increased, and myalgia.
“Since SOD1 mutations were first identified as a cause of ALS 30 years ago, the familial ALS community has been searching for genetically targeted treatments. Qalsody offers families who have lost generation after generation in the prime of their life to this devastating disease a therapy targeting the underlying cause of SOD1-ALS. Today marks an important moment in ALS research as Qalsody is the first ALS treatment approved based on a biomarker,” said Jean Swidler, chair of Genetic ALS & FTD: End the Legacy. “We are excited to see what future therapies are developed now that it is understood that lowering levels of neurofilament provides important evidence that a treatment is affecting the neurodegenerative process.”
The efficacy of Qalsody was assessed in a 28-week randomized, double-blind, placebo-controlled clinical study in patients 23 to 78 years of age with weakness attributable to ALS and a SOD1 mutation confirmed by a central laboratory. One hundred eight (108) patients were randomized 2:1 to receive treatment with either Qalsody 100 mg (n=72) or placebo (n=36) for 24 weeks (3 loading doses followed by 5 maintenance doses). Concomitant riluzole and/or edaravone use was permitted for patients and at baseline 62% of patients were taking riluzone, and 8% of patients were taking edaravone.
Over 28 weeks in VALOR, participants in the primary analysis population (n=60) treated with Qalsody experienced less decline from baseline as measured by the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) compared to placebo, though the results were not statistically significant (Qalsody-placebo adjusted mean difference [95% CI]: 1.2 [-3.2, 5.5]). In the overall intent-to-treat population (n=108), Qalsody-treated participants experienced a 55% reduction in plasma NfL compared to a 12% increase in placebo-treated participants (difference in geometric mean ratios for Qalsody to placebo: 60%; nominal p<0.0001). Additionally, levels of CSF SOD1 protein, an indirect measure of target engagement, were reduced by 35% in the Qalsody-treated group compared to 2% in the corresponding placebo group (difference in geometric mean ratios for Qalsody to placebo: 34%; nominal p<0.0001).