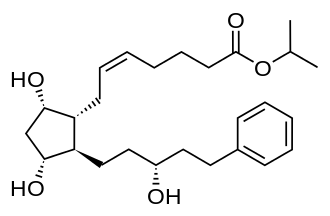
Bausch + Lomb Corporation, announced that the U.S. Food and Drug Administration (FDA) has approved Miebo™ (perfluorohexyloctane ophthalmic solution; formerly known as NOV03), for the treatment of the signs and symptoms of dry eye disease (DED). Miebo is the first and only FDA-approved treatment for DED that directly targets tear evaporation.
“Today’s FDA approval of Miebo further advances DED treatment by addressing a significant unmet need for millions of people suffering with this disease,” said Brent Saunders, chairman and CEO, Bausch + Lomb. “We are proud to bring to market the first and only prescription eye drop approved in the United States for the treatment of DED that directly targets evaporation. We expect to make Miebo commercially available in the second half of this year.”
DED affects millions of Americans and is one of the most common ocular surface disorders.1 A leading cause of DED is excessive tear evaporation, which due to an altered tear lipid layer, is often associated with the clinical signs of Meibomian gland dysfunction (MGD). An unstable tear film triggers increased ocular surface desiccation, inflammation and damage to the ocular surface.2,3 Miebo is designed to reduce tear evaporation at the ocular surface.4,5
In GOBI and MOJAVE, two phase 3 pivotal clinical trials which enrolled more than 1,200 patients (randomized 1:1 to Miebo or hypotonic saline) with a history of DED and clinical signs of MGD, Miebo consistently met its primary clinical sign and patient-reported symptom endpoint.
"In the two pivotal clinical trials, Miebo addressed the persistent and chronic nature of DED by providing sustained improvement in both the signs and symptoms of DED,” said Preeya Gupta, M.D., cornea and cataract surgeon, Triangle Eye Consultants, Raleigh, North Carolina. “Because Miebo inhibits evaporation, it may be an appropriate treatment option for patients whose tear evaporation exceeds tear supply.”
“Tear evaporation, which is a leading driver of DED, presents a significant treatment challenge. With the approval of Miebo, eye care professionals can now take a new approach to DED therapy with a first-in-class water- and preservative-free prescription treatment option that specifically addresses tear evaporation,” said Paul Karpecki, O.D., director, Cornea and External Disease, Kentucky Eye Institute, and associate professor, University of Pikeville, Kentucky College of Optometry.
Ref : Read More